
在生物信息学分析中,PCA(主成分分析)是一种常用的降维与可视化方法,能帮助我们从复杂的基因表达数据中捕捉关键模式,轻松观察样本间的差异与聚类情况。今天,我们就以真实数据为例,一步步教你如何用R语言完成PCA分析与绘图!
关键步骤与实战要点
1、读取数据
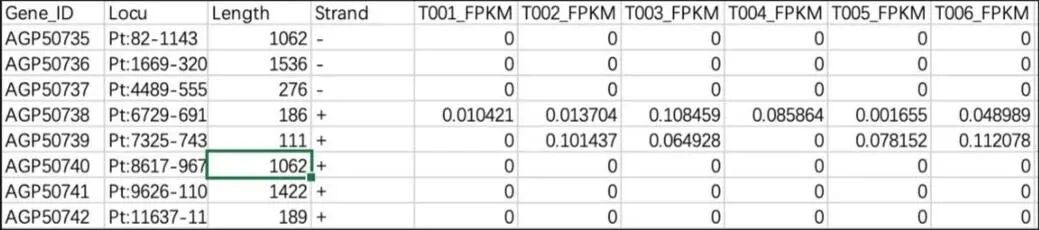
原始数据结构如下图:
第一列为基因ID, 第五列及以后为各个样本的表达量数据,数值以FPKM表示。
读取数据命令如下:
rawdata=
read.csv( ‘AllSample.genes_expression.csv’, header=T)
2、数据清洗
原始数据中包含一些不参与分析的列,以及某些基因的表达量在所有样本中均为零,需要在分析前去除。
操作代码如下:
#将第一列数据作为行名保存
row.names(rawdata)<-rawdata$ Gene_ID
#删除第一列位置信息和第四列正负链的数据,并进行行列转置 tmp<-t(rawdata[,c(-1,-2,-3,-4)])
#删除在所有样本中表达量均为0的基因 cleandata<-tmp[,colSums(tmp!=0)>0]
这样我们就得到了一个“干净”的样本×基因表达矩阵,便于后续分析。
3、PCA 分析
R语言内置的prcomp()函数可以快速完成PCA计算,代码如下:
data.pca< -prcomp(cleandata,center=T, scale.=)
#cleandat 为进行分析的数据集
#center 一个逻辑值,控制变量是否应该移位到零中心
#scale.一个逻辑值,控制是否对数据进行标准化
prcomp 函数的返回值是一个特殊的对象,可以利用summary 函数来查看分析的结果。
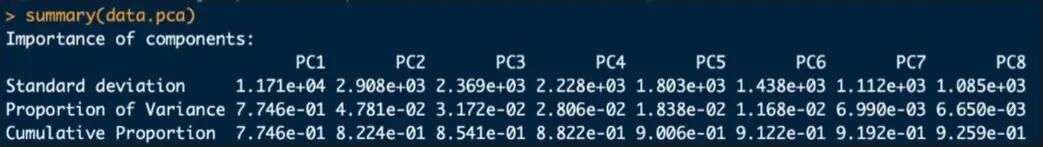
具体参数:
#Standard deviation 标准差,其平方为方差=特征值
#Proportion of Variance方差贡献率
#Cumulative Proportion 方差累计贡献率
输出结果包括各主成分的标准差、方差贡献率等,帮你判断应该保留多少主成分。
4、数据可视化
使用ggplot2包,轻松将PCA结果转换为直观的散点图。
#加载ggplot 软件包 library(ggplot2)
#根据PC1 及PC2生成散点图,由于ggplot在绘图时只接收数据框格式的数据集,因此需要使用as.data.frame函数来进行转换。
ggplot( as.data.frame( data.pcaSx),aes(x=PC1,y=PC2))+ geom_point()
图片生成:
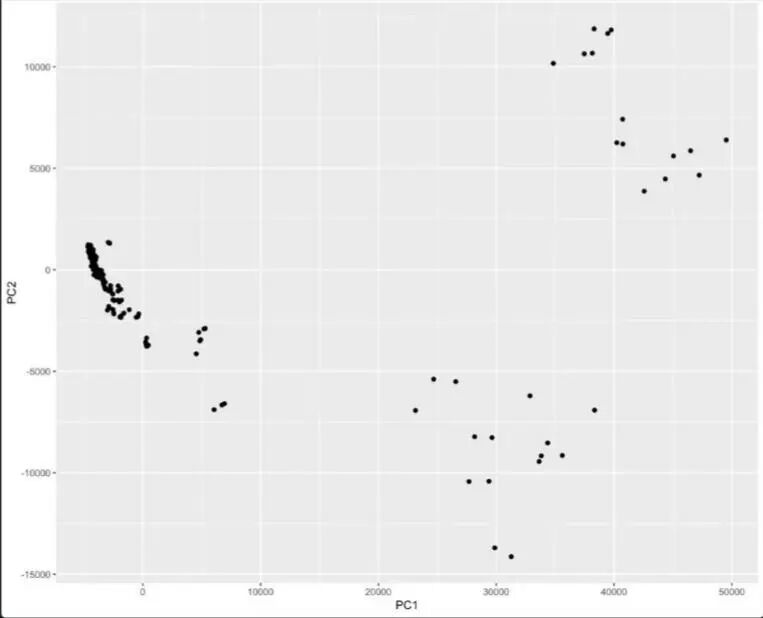
一张清晰的PCA图就诞生了!样本分布、组间差异一目了然。
核心要诀与进阶之路
PCA图不仅能展示样本聚类,还能通过颜色、形状区分不同实验组,助力发现潜在离群样本或批次效应,是表达数据分析的必备技能!如想学习更多生信分析技能,请登录百迈客生物云课堂知识库,在线学习更多生信分析技巧。




 京公网安备 11011302003368号
京公网安备 11011302003368号