

2026年4月1日,肝病学国际知名期刊Journal of Hepatology在线发表了苏州大学、复旦大学、南方科技大学等单位联合完成的重磅研究成果:“Chromatin remodeling in pericentral hepatocytes modulates MASH through CYP450 activity”。

该研究突破性地揭示了肝小叶中央静脉周围Lgr5+肝细胞中,染色质重塑因子DPF2通过CYP450-全反式维甲酸(atRA)-AMPK信号轴,驱动MASH发生发展的核心机制,为MASH的早期诊断和靶向治疗开辟了全新方向。百迈客生物为该研究提供了百创空间转录组BMKMANU S3000测序服务。
研究背景
代谢相关脂肪性肝病(MASLD)已成为全球慢性肝病的首要病因,全球患病率超30%,进展为代谢相关脂肪性肝炎(MASH)的患者逐步发展为肝纤维化、肝硬化甚至肝细胞癌,是全球肝病领域亟待攻克的重大公共卫生难题。截至目前,MASH的发病机制尚未完全阐明,临床仍缺乏精准的早期诊断标志物和特效治疗药物。
肝脏是具有高度空间分区特征的器官,肝小叶从门静脉到中央静脉被划分为不同功能分区,不同区域的肝细胞在基因表达、代谢功能上存在显著异质性。既往针对MASLD/MASH的研究多采用全肝bulk测序或单细胞测序,无法还原关键致病细胞亚群的原位空间定位与分区调控特征,难以解释“少量特殊肝细胞亚群驱动全肝代谢紊乱”的核心谜题,成为该领域长期难以突破的技术瓶颈。
研究结果
Lgr5+肝细胞在肝脏稳态与MASH进展中保持稳定,Dpf2缺失显著加剧MASH表型
作者首先构建了Lgr5-CreERT2;H11-CAG-LSL-tdTomato谱系示踪小鼠模型(图1A),通过CDAA饮食诱导MASH模型,明确了核心研究对象的基本特征:在MASH进展的4周周期内,小鼠肝脏脂质沉积随饮食喂养时间持续加重,但Lgr5+肝细胞占比始终稳定在5%左右,不随疾病进展发生显著变化(图1B-C),证实该细胞群在肝脏稳态和MASH进程中持续存在,是潜在的疾病调控关键亚群。
基于肥胖患者scRNA-seq数据集的富集分析,作者锁定了SWI/SNF染色质重塑复合物的核心亚基Dpf2作为研究靶点,构建了Lgr5+肝细胞特异性Dpf2敲除小鼠(Dpf2ΔLgr5)(图1D)。功能实验结果显示:在CDAA饮食诱导下,Dpf2ΔLgr5小鼠的肝脏脂质沉积、血清ALT/AST水平(肝损伤核心指标)均显著高于野生型(WT)对照组,MASH病理表型显著加剧(图1E-G);反之,通过AAV8-DIO-Dpf2体系在Lgr5+肝细胞中特异性过表达Dpf2,可显著缓解CDAA饮食诱导的MASH损伤(图1H-K),首次证实Lgr5+肝细胞中的Dpf2是MASH进展的关键保护性调控因子。
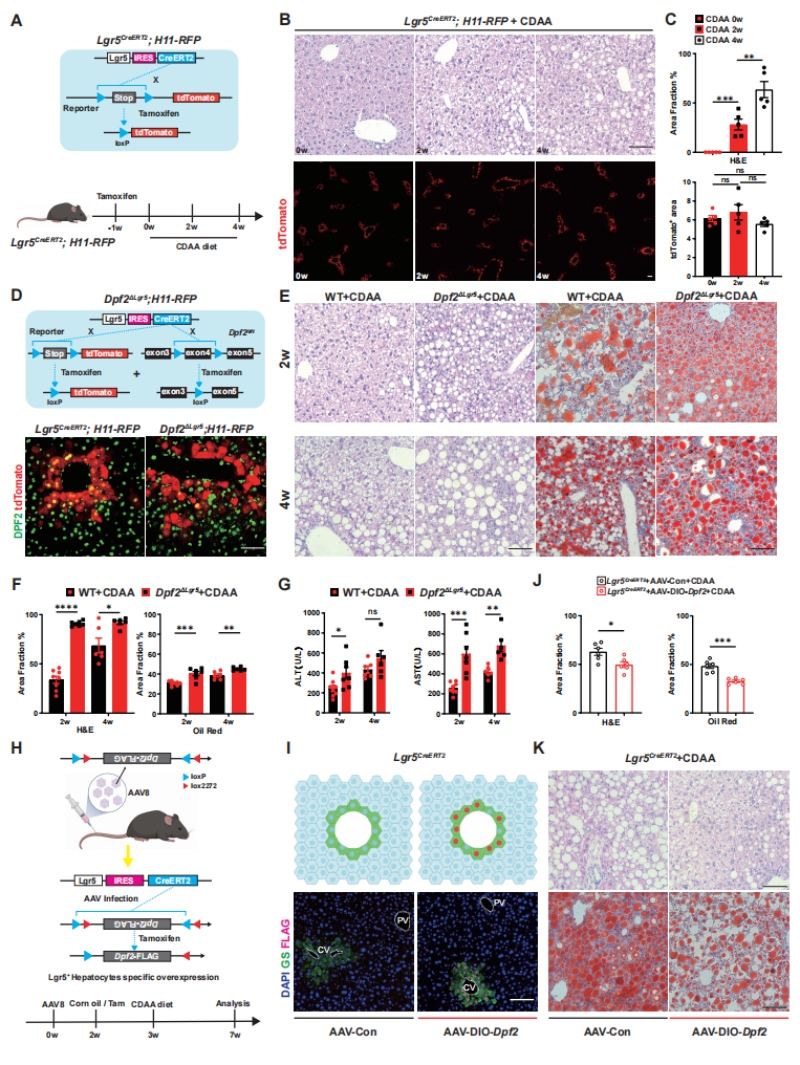
图1
Lgr5+肝细胞中Dpf2缺失显著加速MASLD进展
为进一步验证Dpf2在MASLD中的调控作用,作者采用FPC饮食(果糖-棕榈酸-胆固醇饮食)诱导更贴合临床特征的MASLD模型,发现FPC饮食喂养28天后,Dpf2ΔLgr5小鼠肝脏体积、肝重/体重比显著升高,肝脏脂质沉积、炎症因子(Tnfa、Ccl2)和纤维化相关基因(Col1a1、Col3a1)的表达水平均显著高于WT对照组,肝损伤指标ALT/AST持续升高(图2A-G)。
病程对比实验证实,Dpf2ΔLgr5小鼠喂养28天的MASLD严重程度,超过了WT小鼠喂养12周的表型,证实Dpf2缺失可使MASLD进展速度加快超8周;肝细胞靶向AAV回补实验排除了肠道Lgr5+干细胞的干扰,证实仅在Lgr5+肝细胞中回补Dpf2,即可完全逆转Dpf2ΔLgr5小鼠的严重MASLD表型(图2H-I),明确了Lgr5+肝细胞中的Dpf2是肝脏代谢稳态的特异性、非冗余调控因子。
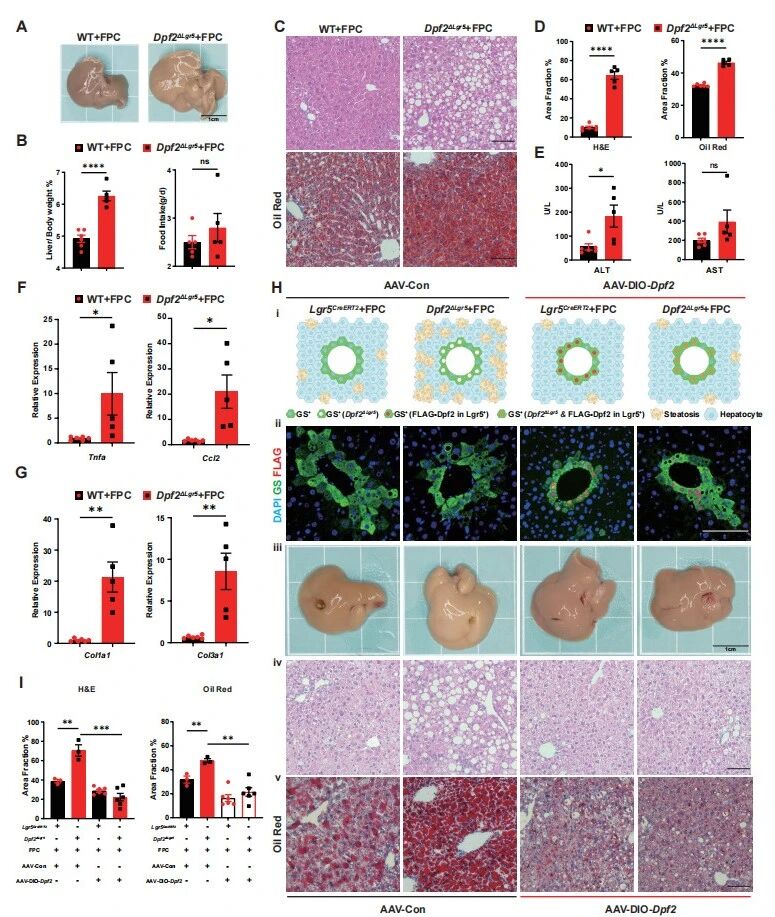
图2
Lgr5+肝细胞通过代谢物atRA实现与全肝肝细胞的非细胞自主通讯
针对Lgr5+肝细胞如何调控全肝代谢的核心科学问题,作者整合了scRNA-seq与snRNA-seq数据,构建了包含88223个细胞的肝脏单细胞转录组图谱,注释得到10种主要细胞类型,并对肝细胞完成了分区亚群细分(图3A)。
通过细胞通讯分析,作者首次发现Lgr5+肝细胞可通过全反式维甲酸(atRA)与肝小叶1区、3区的肝细胞完成跨区域通讯(图3B);代谢组学与质谱检测证实,Dpf2ΔLgr5小鼠肝脏中atRA的前体视黄醇、以及成熟atRA的含量均发生显著耗竭(图3C-D)。
功能回补实验验证了该通路的核心作用:给FPC饮食喂养的小鼠外源性补充atRA,可显著缓解肝脏脂质沉积、炎症与纤维化表型,同时逆转Dpf2缺失导致的MASLD损伤(图3E-H),证实Lgr5+肝细胞正是通过调控atRA的代谢稳态,实现对全肝代谢的非细胞自主调控。
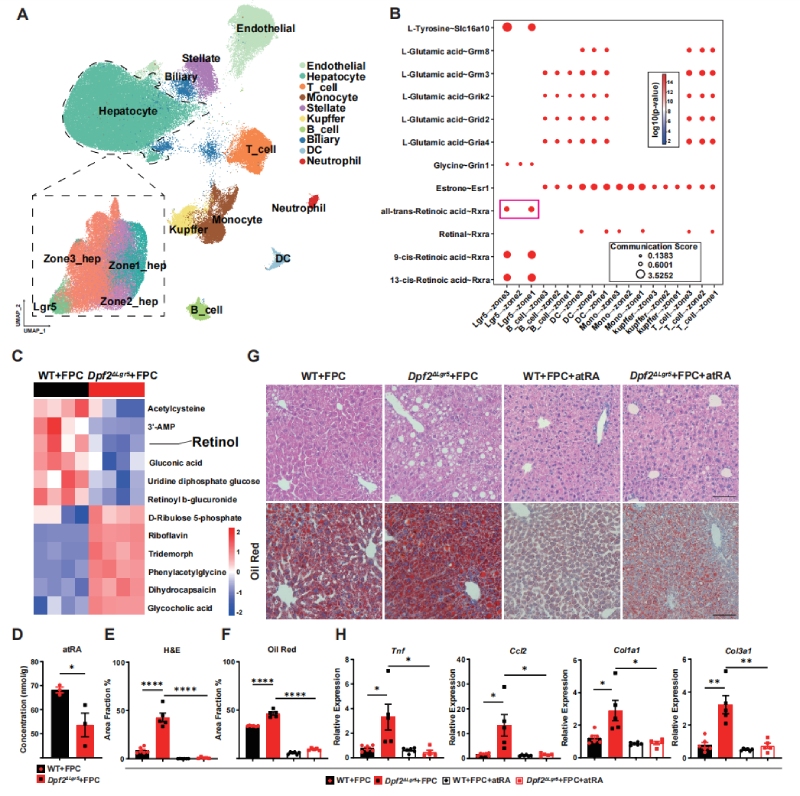
图3
百创S3000空间转录组揭示Dpf2缺失通过激活Cyp2s介导的视黄醇分解代谢通路驱动疾病
为在原位空间层面解析atRA耗竭的分区调控机制,作者采用百创S3000空间转录组测序平台(BMKMANUS3000),对FPC饮食喂养的WT与Dpf2ΔLgr5小鼠肝脏组织进行了高分辨率空间转录组分析。精准完成肝脏组织的空间细胞分群,注释出肝细胞的4个分区亚群(门静脉区zone1、中间区zone2、中央静脉周围zone3/PC区、Lgr5+肝细胞亚群),并验证了Dpf2缺失不影响肝细胞的整体增殖能力(图4A-B)。
空间基因集富集分析(GSEA)证实,Dpf2ΔLgr5小鼠肝脏的中央静脉周围(PC)区,视黄醇代谢通路发生了最显著的上调,与scRNA-seq中Lgr5+肝细胞的通路富集结果完全一致(图4E-F)。百创S3000的空间表达可视化结果明确显示:负责atRA分解代谢的CYP450家族2亚家族(Cyp2s)基因,在Dpf2ΔLgr5小鼠的中央静脉周围区发生了特异性、显著的表达上调,与Lgr5+肝细胞的空间分布高度重合(图4G-H);单细胞水平的表达分析也证实,Cyp2s基因的上调主要集中在Lgr5+肝细胞与zone3肝细胞中(图4I)。
免疫组化验证与临床转化分析同步完成:小鼠肝脏IHC染色证实Dpf2ΔLgr5小鼠中央静脉周围区的CYP2C9、CYP2C19(Cyp2s的人源同源基因)蛋白水平显著升高(图4J);人源脂肪肝临床样本的H&E、IHC染色,以及公共数据库中人源MASLD样本的空间转录组数据分析,均证实脂肪肝患者肝脏中存在CYP2s基因的表达上调与视黄醇代谢通路的异常激活(图4K-P),完成了从动物模型到人类疾病的转化验证。
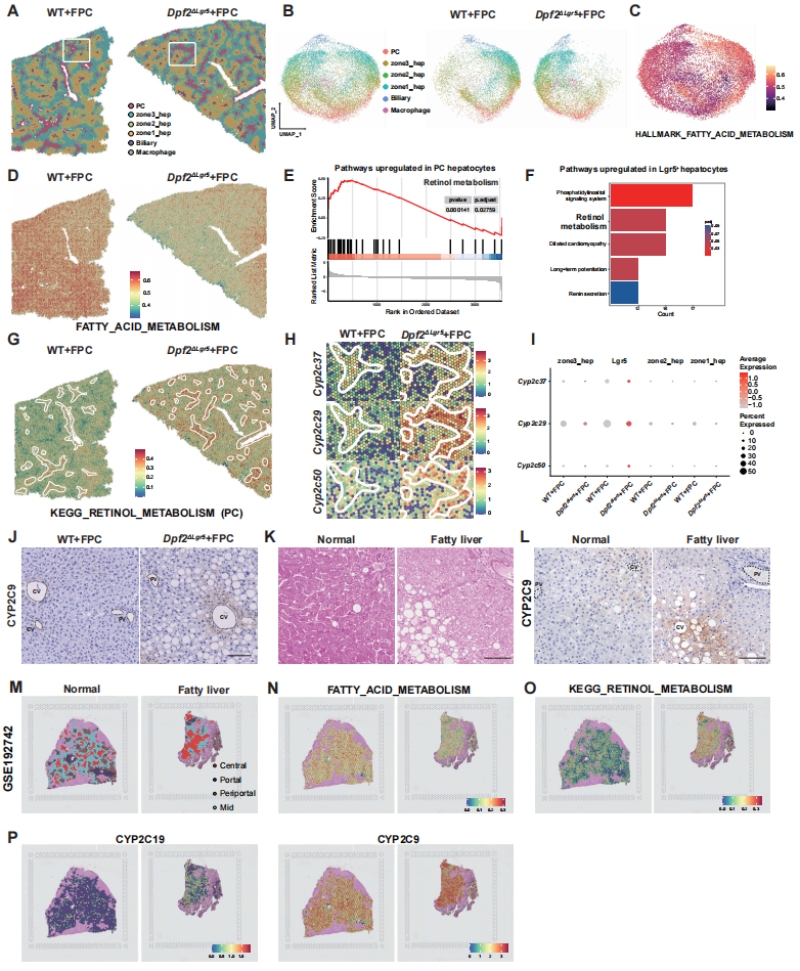
图4
Dpf2通过表观遗传调控直接抑制Cyp2s基因的转录激活
为明确Dpf2调控Cyp2s基因表达的分子机制,作者通过流式分选获得了WT与Dpf2ΔLgr5小鼠的Lgr5+肝细胞,结合ATAC-seq、CUT&Tag、CUT&RUN等表观基因组技术,完成了调控机制的解析(图5A)。IGV可视化与全基因组分析结果显示,Dpf2缺失后,Lgr5+肝细胞中Cyp2s基因(Cyp2c50、Cyp2c29、Cyp2c37)的转录起始位点(TSS)区域,染色质可及性(ATAC-seq信号)与激活型组蛋白修饰H3K4me3水平均显著升高,证实Dpf2缺失可直接开放Cyp2s基因的染色质状态,驱动其转录激活(图5B-D)。
功能抑制实验验证了该靶点的治疗价值:给Dpf2ΔLgr5小鼠每日灌胃CYP酶强效抑制剂柚皮苷(Naringin),可显著缓解FPC饮食诱导的肝脏脂质沉积、炎症与纤维化表型,逆转MASLD进展(图5E-I),证实Cyp2s是Dpf2下游调控MASH进展的直接功能靶点。
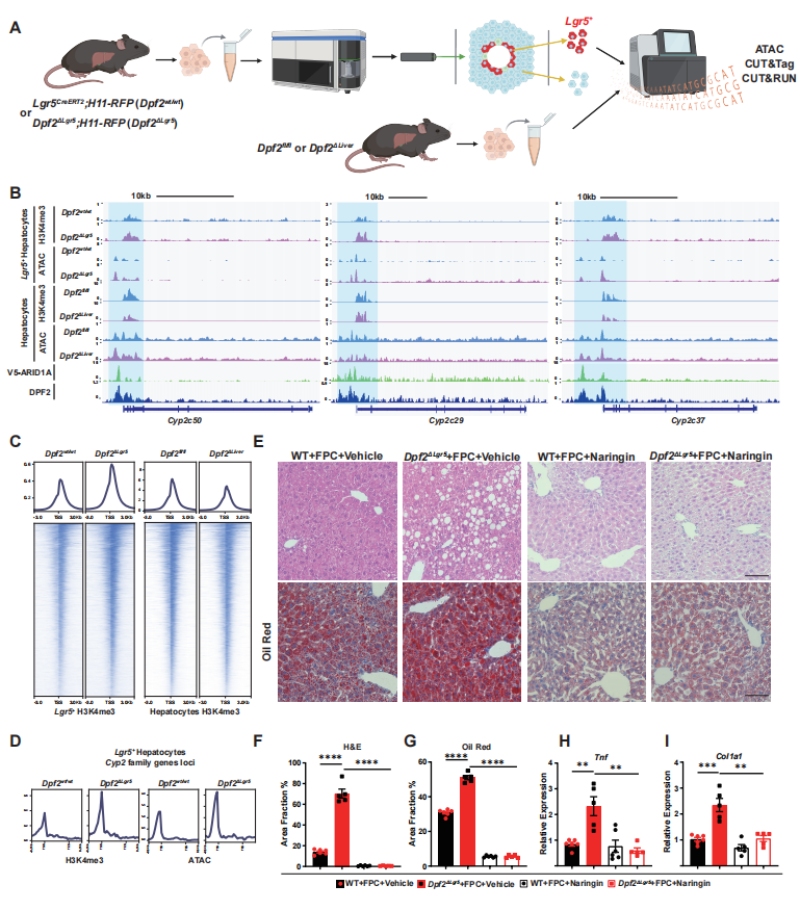
图5
atRA通过激活AMPK信号通路发挥肝脏保护作用
作者进一步解析了atRA调控全肝代谢的下游分子机制,通过空间转录组、scRNA-seq的联合分析发现,Dpf2ΔLgr5小鼠肝脏中AMPK信号通路发生显著下调,AMPK的靶基因Fasn(脂质合成关键基因)显著上调、Mlycd(脂肪酸氧化关键基因)显著下调,且该变化遍及全肝所有肝细胞分区(图6A-D)。
通过体内功能获得与缺失实验,作者完整验证了atRA-AMPK轴的核心作用:通过尾静脉水动力注射组成型激活的AMPK(CA-AMPK)质粒,或使用AMPK激动剂A-769662干预,均可显著逆转Dpf2ΔLgr5小鼠的严重MASLD表型;采用zone1/2肝细胞特异性启动子驱动的AAV-Gls2-CA-AMPK靶向回补,同样可显著缓解肝脏脂质沉积,证实AMPK是atRA下游的核心效应分子(图6E-K)。在小鼠肝脏中过表达显性负突变的AMPK(DN-AMPK),可完全阻断atRA对MASLD的保护作用,证实atRA的肝脏保护效应完全依赖于AMPK信号通路的激活(图7A-E)。
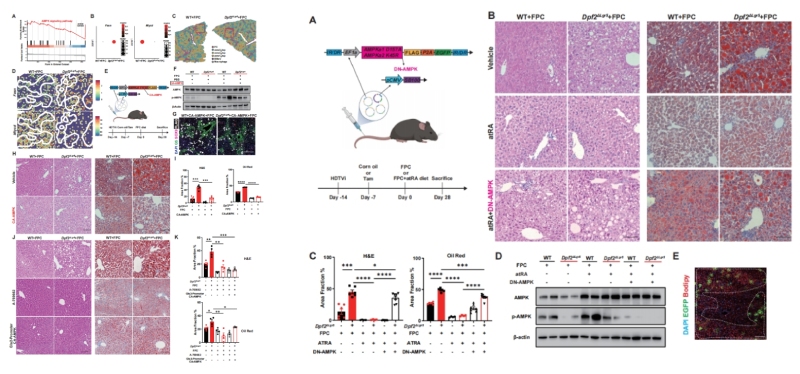
左:图6 、 右:图7
研究总结
该研究突破性地揭示了肝小叶中央静脉周围仅占5%的Lgr5+稀有肝细胞亚群,通过SWI/SNF染色质重塑复合物亚基Dpf2,以表观遗传方式调控CYP2s-atRA-AMPK信号轴驱动全肝代谢紊乱与MASLD/MASH进展的全新机制。不仅刷新了领域对肝脏分区代谢调控、非细胞自主代谢调控的基础认知,破解了长期以来“少量肝细胞亚群驱动全肝疾病”的核心谜题,更为全球高发的代谢相关脂肪性肝病提供了全新的早期诊断标志物与精准治疗靶点,具有里程碑式的科学价值与临床转化意义。
百迈客生物自主研发的百创S3000空间转录组测序平台提供技术支撑:该平台凭借2.5μm亚微米级的超高捕获分辨率,完美适配肝小叶高度分区化的精细结构特征,实现了对门静脉区到中央静脉区肝细胞亚群的精准空间分群;同时,百创S3000标准化的全流程实验体系,为研究提供了稳定、高质量、可重复的顶刊级测序数据。
文献引用:10.1016/j.jhep.2026.03.035 Chromatin remodeling in pericentral hepatocytes modulates MASH through CYP450 activity




 京公网安备 11011302003368号
京公网安备 11011302003368号