
全长转录组测序(Nanopore平台)
产品介绍
Nanopore全长转录组测序是指基于牛津纳米孔公司(Oxford Nanopore Technologies,ONT)三代测序平台进行全长转录组测序,无需打断,可直接读取从5’端到3’端polyA尾的高质量单个RNA分子全长序列,准确辨别二代测序无法准确识别的可变剪接(AS)、可选择性多聚腺苷酸化(APA)、融合基因、lncRNA及其靶基因,且可同时对基因和转录本进行定量分析。ONT全长转录组已广泛应用于生长发育、环境适应、免疫互作、突变表型、肿瘤的发生、临床诊断和药物研发等领域。
技术路线
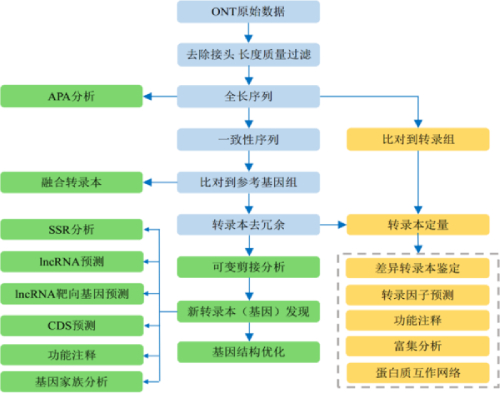
应用方向
结果展示
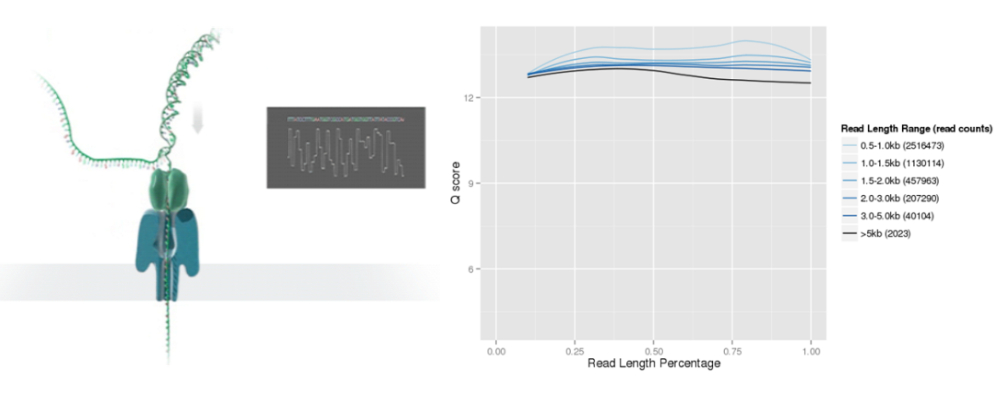
数据质控
Nanopore测序通过对纳米孔的DNA/RNA单分子实时电信号进行检测及对应,即可计算获得相应碱基的类型,完成序列的实时测定。为确保Reads有足够高的质量,将下机原始测序数据(raw reads)过滤短片段和低质量的reads,得到clean reads,保证后续分析的准确性。
将reads按照长度从短到长排序,平均分成10份,每一份统计reads的平均质量值,平均质量值Q10以上,即为合格。
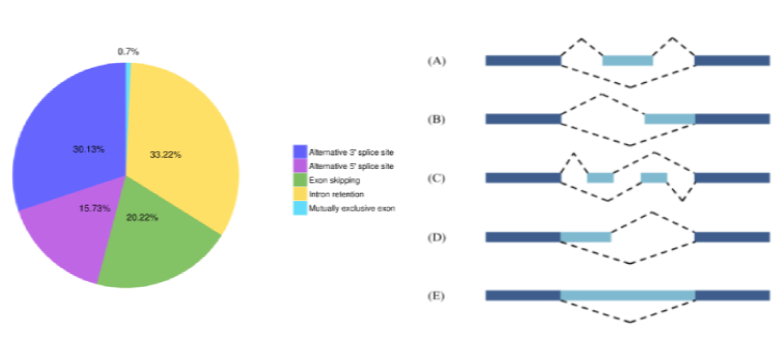
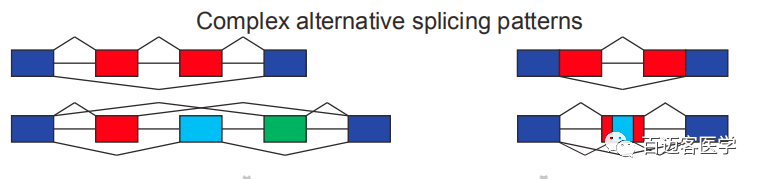
可变剪切鉴定
基因转录生成的前体mRNA(pre-mRNA),有多种剪接方式,选择不同的外显子,产生不同的成熟mRNA,从而翻译为不同的蛋白质,构成生物性状的多样性。这种转录后的mRNA加工过程称为可变剪接或选择性剪接(Alternative splicing)。可变剪接类型包括:(A) 外显子跳跃;(B) 可变转录终止位点;(C) 可变外显子;(D)可变转录起始位点;(E) 内含子保留。
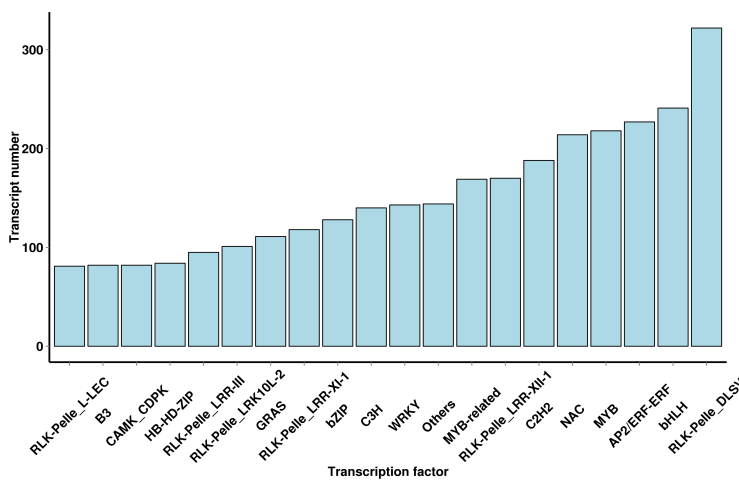
转录因子预测
转录因子(Transcription factor)是指能够结合在某基因上游特异核苷酸序列上的蛋白质,这些蛋白质可以调控RNA聚合酶与DNA模板的结合,从而调控基因的转录。
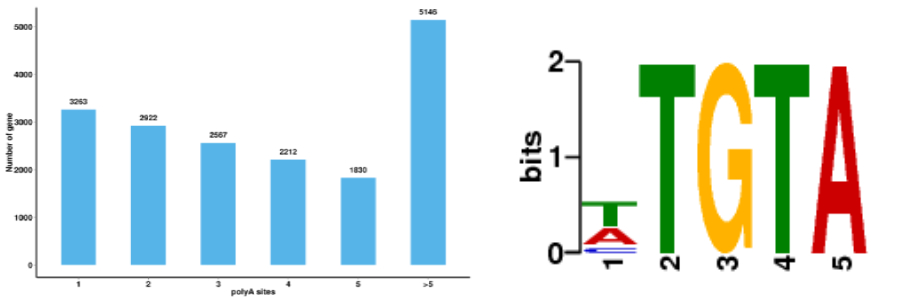
可选择性多聚腺苷酸化分析-APA
多聚腺苷酸化是指多聚腺苷酸与信使RNA(mRNA)分子的共价链结。在蛋白质生物合成的过程中,这是产生准备作翻译的成熟mRNA的方式的一部份。在真核生物中,多聚腺苷酸化是一种机制,令mRNA分子于它们的3’端中断。多聚腺苷酸尾(或聚A尾)保护mRNA,免受核酸外切酶攻击,并且对转录终结、将mRNA从细胞核输出及进行翻译都十分重要。前体mRNA的可变多聚腺苷酸化(alternative polyadenylation, APA)可能贡献于转录组多样性,基因组的编码能力以及基因的调控机制。
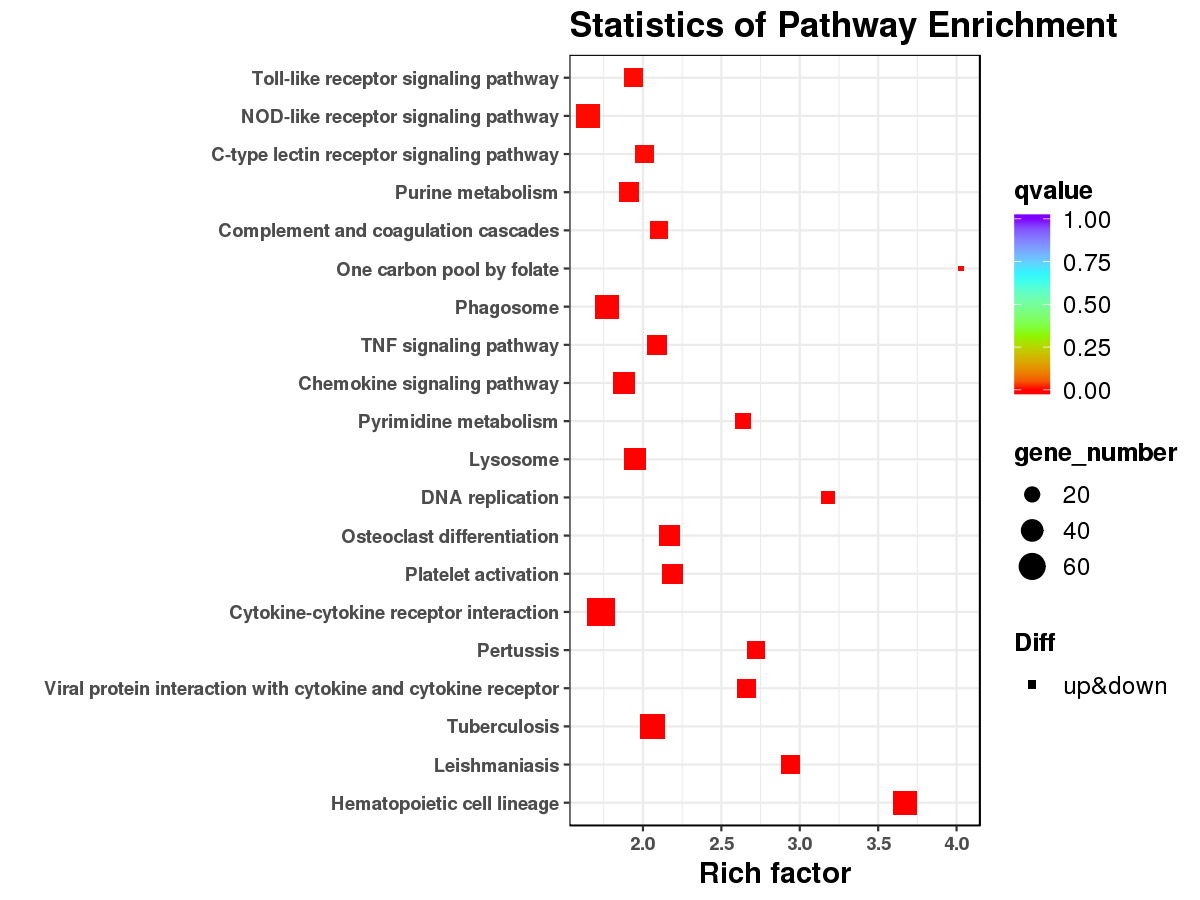
差异表达基因/转录本KEGG通路富集
KEGG Pathway富集可以系统分析基因产物在细胞中的代谢途径以及这些基因产物功能,把基因/转录本及表达信息作为一个整体的网络进行研究。
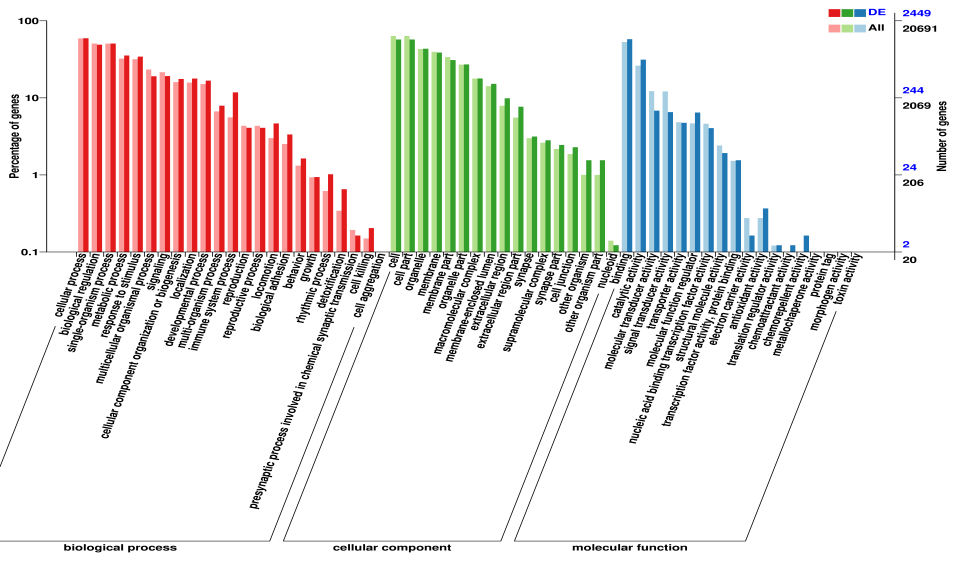
差异表达基因/转录本GO注释
GO注释分类统计图,直观的反映出在生物过程(biological process)、细胞组分(cellular component)和分子功能(molecular function),可深入挖掘差异基因和转录本的功能及所在的信号通路,筛选关注差异基因和转录本注释情况。
常见问题
什么是nanopore全长转录组测序?
nanopore全长转录组测序是指基于牛津纳米孔公司(Oxford Nanopore Technologies,ONT)三代测序平台进行全长转录组测序。全长转录组测序,无需打断,基于三代测序平台直接获取转录本的5ˊ到3ˊ高质量全长序列,可准确识别可变剪接、基因融合、可选择性多聚腺苷酸化APA、等位基因特异性表达等转录本结构方面变异。基于nanopore三代测序平台进行全长转录组测序,除了可准确鉴别上述转录本结构变异,还可实现转录本(mRNA或polyA+ lncRNA)表达水平准确定量。
nanopore全长转录组测序与二代Illumina平台普通转录组的区别?
主要在于测序平台不同。Illumina平台主要是PE150测序,构建小片段文库,为边合成边测序,在建库以及测序过程中均需要PCR扩增,主要用于基因水平表达定量及差异表达分析。nanopre全长转录组测序无需打断RNA,可获得5’到3’全长转录本序列及其表达信息,对片段大小无偏好,直接检测电信号无需边合成边测序其GC偏好性远低于二代平台;同时由于无需拼接其在转录本层面的结构变异检测方面,比如可变剪接、融合基因、APA、新基因预测等具有绝对优势。
nanopore全长转录组测序和二代转录组测序平台有何差异?
nanopore测序是基于电信号识别碱基序列的三代测序技术。DNA/RNA上不同碱基或带不同修饰时化学性质存在差异,当单链分子通过纳米孔通道时,碱基造成的阻碍大小不一,因此会形成特征性离子电流变化信号。通过对这些信号进行实时检测,即可获得相应碱基类型,完成测序。目前通过“递归神经网络(Recurrent Neural Network)”的复杂算法对碱基进行判读。
其特点为:
1)读长长:最长读长能达到2 Mb以上级别[ref1],有利于可变剪接、基因融合等结构变异检测;
2)低成本:相比其他三代测序技术,ONT测序样本处理极其简单,无需DNA聚合酶、连接酶和dNTPs,测序价格低;
3)测序过程不涉及PCR扩增:避免二代测序中PCR扩增可能引入的错误或丰度变化;
4)direct-RNA/DNA方式建库,可直接获取碱基修饰信息,如甲基化修饰5mC、6mA等,无须像二代测序需要经过重硫酸盐转化或者免疫沉淀富集实验;
5)低GC含量和碱基偏好性,针对RNA测序无需打断,转录本水平表达定量更准确。
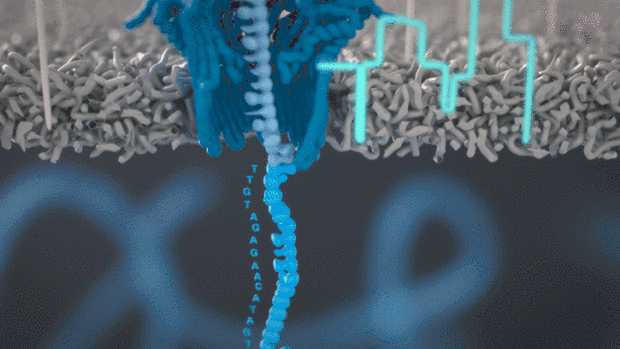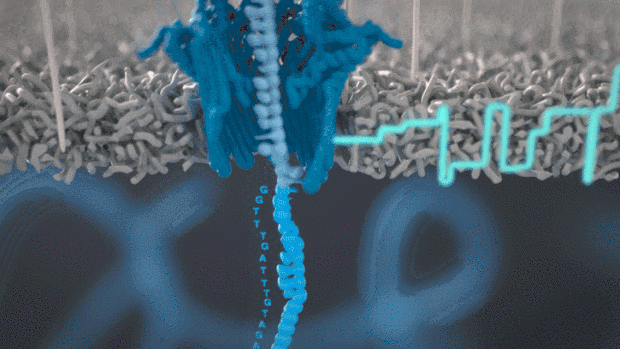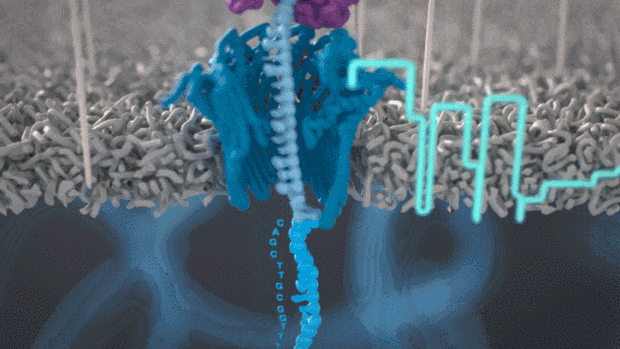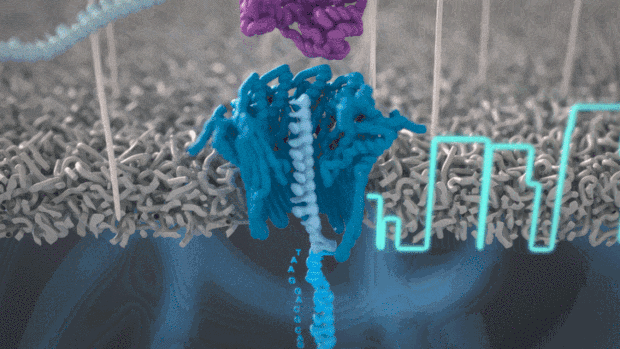
二代转录组测序一般基于边合成边测序二代短读长的Illumina等平台,由于读长短(通常PE 150bp),需要进行片断化,打断到200-300bp,测序过程需要DNA聚合酶和dNTPs以及进行桥式PCR形成clusters放大荧光信号。
从二代转录组到nanopore三代全长转录组,平台升级,技术革新,解决二代不能解决的问题!
nanopore全长转录组测序送样要求?
样品类型:PolyA RNA;样品浓度:≥50 ng/ul(Qubit HS RNA定量); 样品总量:cDNA-direct方式:>250ng(单次);总量>750 ng;(若提供总RNA,动物样品总量需按照PolyA RNA要求的100倍以上准备); cDNA-PCR方式:>1μg(单次);总量>3μg 样品纯度:OD260/280 ~2.0,OD260/230在2.0-2.2 之间,260nm处有正常峰值;样品无基因组DNA污染; 总RNA完整性: RIN值≥8.0,28S/18S≥1.0;图谱基线无上抬;5S峰正常。
nanopore全长转录组测序一般建议多少个生物学重复?
研究表明,生物学重复可提高所有基因表达水平鉴定的准确性,而增加测序深度主要提高低表达基因表达量鉴定准确性。每种处理条件下至少3个生物学重复,当研究样本的生物学差异比较高,或者想研究更多的微小表达差异/fold change时,需要更多生物学重复。也就是,比如对于个体差异较大的临床样本可以5-10个/组以上,而生物学差异较小的细胞系样本则每组3个生物学重复以上即可。
为什么说nanopore全长转录组测序比二代转录组测序在转录本表达水平定量更准确?
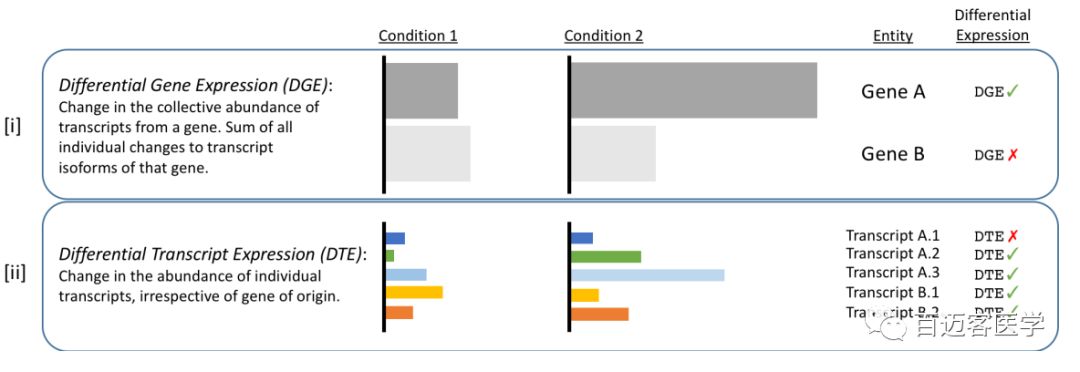
- 首先,转录本表达定量,是指针对一个基因的多个不同转录本(transcript)分别进行表达定量。而基因水平定量,可以理解为一个基因所有转录本表达定量的加和。
- 多数公司二代转录组测序不提供一个基因的多个转录本分别定量的结果,原因在于二代转录组测序并适用于这种情况。二代短测序单条read无法跨越全长转录本,其表达定量在reads比对完成后需要用诸如StringTie等软件进行短reads组装拼接得到转录本再进行其表达量评估。
- 那么,二代转录组测序可能会存在拼接错误或者是拼接不完整,不能准确获得完整转录本,导致各个转录本水平表达定量不准确。
- 尤为重要的是,二代测序对于一个基因的多个转录本得到的reads,尤其是多个转录本共享的外显子区对应reads无法区分其来源转录本,导致转录本水平定量不准确。
- 还有一些情况,当不同基因具有比较相似的高度保守区时,这些区域的短reads也无法准确区分其来源基因/转录本,即存在多比对的现象,这也导致转录本甚至基因水平表达定量存在偏倚。
- nanopore三代全长测序无需对各转录本打断和拼接,一条read即可跨越全长转录本,多比对率也比较低,因此能够准确获取一个基因的多个全长转录本各自的表达信息。
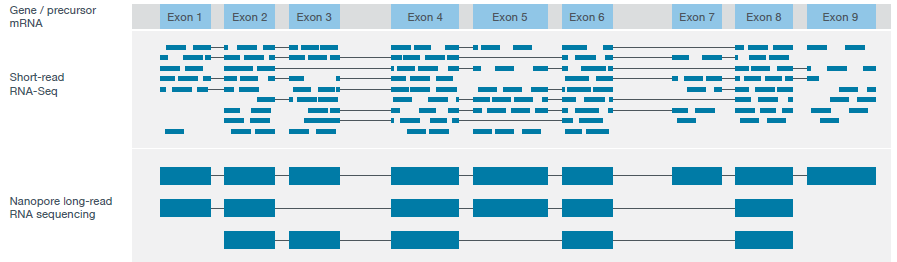 注:上图中蓝色方块表示测到RNA序列中能够比对到基因组序列的区域,即属于某个转录本外显子区的reads;灰色线条表示我们测到的reads回比基因组序列时中间没有比对上的,则用灰色线条连接,即reads不连续比对,中间被剪接掉的内含子区。从图中明显看出,二代短reads比较短,多数短read单条连一个外显子区都跨越不了,nanopore长读长测序可以直接得到3种全长转录本。其中,Exon2和Exon6以及Exon9是三个转录本间共享的,那么对于完全比对到这3个外显子区的短reads无法区分其来源转录本。而三代测序则直接跨越三个全长转录本,因此对于转录本水平表达定量更准确。
注:上图中蓝色方块表示测到RNA序列中能够比对到基因组序列的区域,即属于某个转录本外显子区的reads;灰色线条表示我们测到的reads回比基因组序列时中间没有比对上的,则用灰色线条连接,即reads不连续比对,中间被剪接掉的内含子区。从图中明显看出,二代短reads比较短,多数短read单条连一个外显子区都跨越不了,nanopore长读长测序可以直接得到3种全长转录本。其中,Exon2和Exon6以及Exon9是三个转录本间共享的,那么对于完全比对到这3个外显子区的短reads无法区分其来源转录本。而三代测序则直接跨越三个全长转录本,因此对于转录本水平表达定量更准确。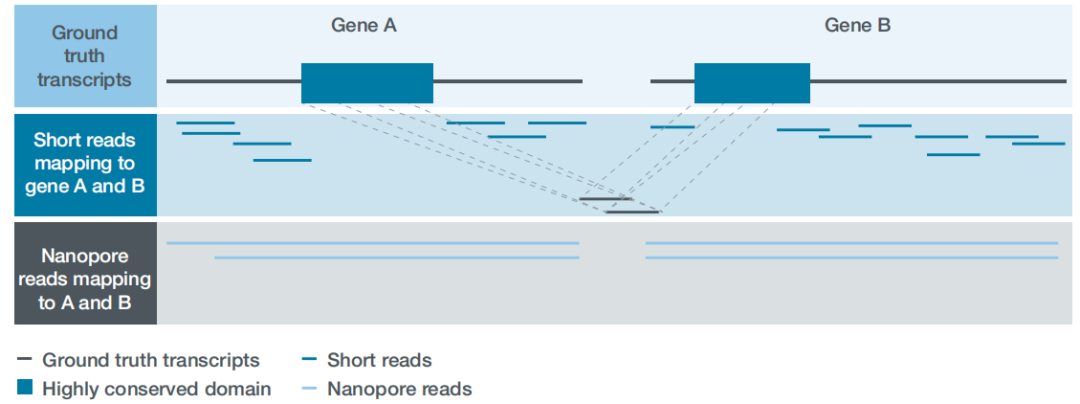
——图片来源于Oxford Nanopore Technologies官方白皮书The value of full-length transcripts without bias。
- 来自同一基因的多个转录本可能行使不同功能,二代测序由于片段化测序导致对转录本水平定量不准确,对于转录本水平表达定量存在固有缺陷,nanopore三代测序弥补了此方面不足,尤其是对于基因水平不显著差异表达的基因,该基因的某些转录本可能显著差异表达,这些转录本可能具有重要生物学作用。比如有些基因的某些转录本只在特定条件下才表达。如果不能对转录本水平进行准确定量,则可能遗漏这些可能具有重要生物学功能的转录本。
为什么说nanopore全长转录组测序比二代转录组测序在AS、APA、基因fusion等结构变异以及复杂转录本鉴定方面更准确?
- 人类中约95%基因存在可变剪接AS事件、70%以上基因存在APA现象[ref3]。AS、APA和fusion等是转录本多样性以及蛋白多样性的来源,多样性转录本或蛋白发挥不同功能。
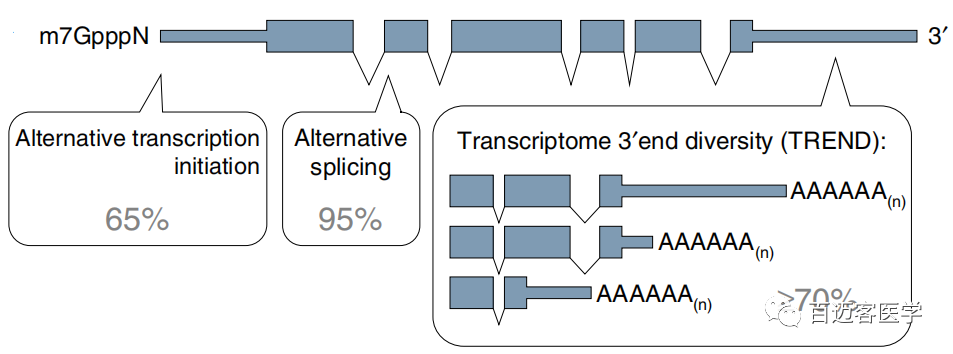
- 由于全长转录本序列的获得,使得nanopore全长转录组测序在鉴定可变剪接(alternative splicing, AS)、可选择性多聚腺苷酸化(APA)以及融合转录本(fusion transcripts)等转录本层面的结构变异更准确,这是之前三代长读长测序广为熟知的应用。
- 目前利用二代Illumina高通量测序方法进行可变剪切的检测与分析非常普遍,但由于二代测序的读长短,在准确预测完整的isoform全长序列方面存在问题,同时存在难以判断转录起始位点(TSS)和转录终止位点(TTS)的位置、难以判断哪些外显子是连接在一起的问题。
- 针对同时发生多个外显子跳跃和内含保留等复杂转录本鉴定更是无能为力。
- 利用二代短序列测序来检测融合基因存在一些问题:基因组重复序列以及多比对率比较高使得检测结果不准确。
- 二代测序技术中为了研究APA已经开发了不下于10种实验技术方法,比如polyA-seq,虽然其可以提取准确的polyA位置,但oligo(dT)引物可能结合到转录本内部的连续A序列,导致假阳性率高,后续需要RNA Pol II结合等额外实验验证这些连续A序列确实是由APA引起的;而如果不进行专门的polyA-seq,普通的二代转录组测序对APA检测效能低下,只能鉴定到约0.003%含有polyA的reads,并且短3’UTR通常嵌在长的UTR中,因此具有短3’UTR的isoform通常被转录组装工具忽视。[ref5]
- nanopore三代全长测序无需对各转录本打断和拼接,poly(A)会出现在测序结果中,并且可以得到从5’到3’的完整全长转录本,其多比对率也比较低,因此能够准确鉴定转录本水平各结构变异。
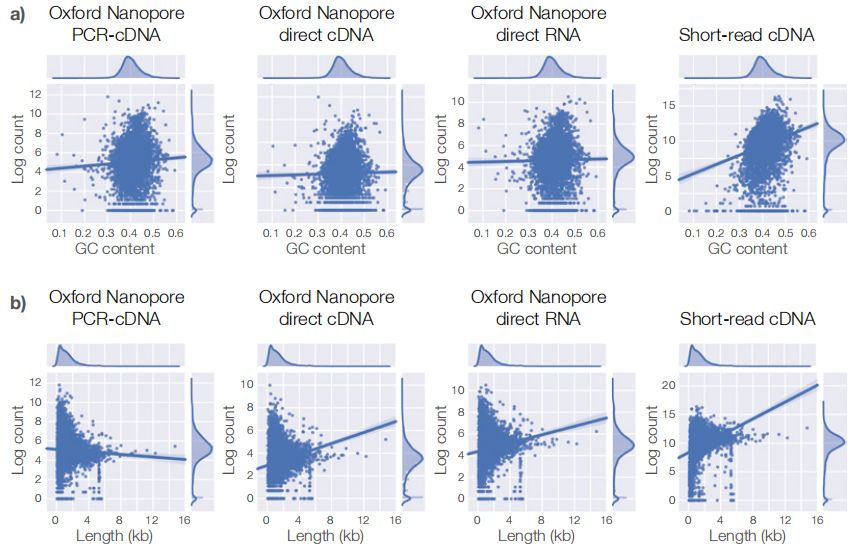
基因的GC含量和长度偏好影响基因表达定量结果,那么nanopore全长转录组测序比二代转录组测序在GC含量和长度偏好方面如何?
使用三种纳米孔建库方式(PCR-cDNA、direct-cDNA和direct-RNA)数据和典型的短读长cDNA技术制备酵母转录组文库进行比较:
a)在所有情况下,纳米孔长读长数据集的GC偏好都比短读长数据集低。
b)与短读长测序数据相比,纳米孔长读长测序数据的长度偏倚都较小。
综上,nanopore全长转录组测序受基因的GC含量和长度偏好更小。
——图片来源于Oxford Nanopore Technologies官方白皮书The value of full-length transcripts without bias。
nanopore平台相比二代平台测序错误率高,那么ONT全长转录本的定量是否可靠?
ONT平台目前我司下机数据碱基质量Q值平均约在10左右,即碱基平均错误率为10^(-1)=10%左右,但这是单碱基错误率;
比对时用的是全长序列和参考基因组或参考转录组进行比对,序列越长比对时对于碱基错配度容忍越高,因此不会对表达定量有影响;
当然由于比二代单碱基错误率高,故而百迈客将SNP和InDel检测分析内容去除了,因此想从RNA水平检测snp/indel的客户可能考虑做二代转录组测序。其实目前不乏使用nanopore测序数据检测snp的文章,如NC|nanopore全基因组重测序鉴定人类基因组非同义新生SNP。
下面列举了2个百迈客真实项目的数据质量表,大家可以参考。
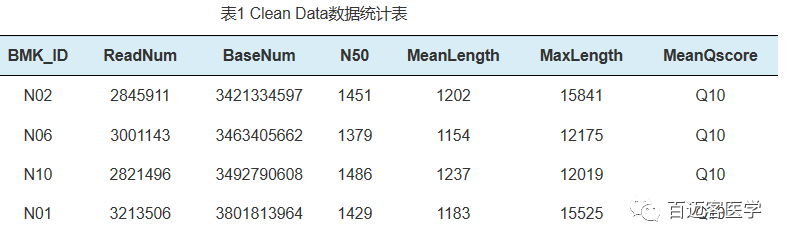
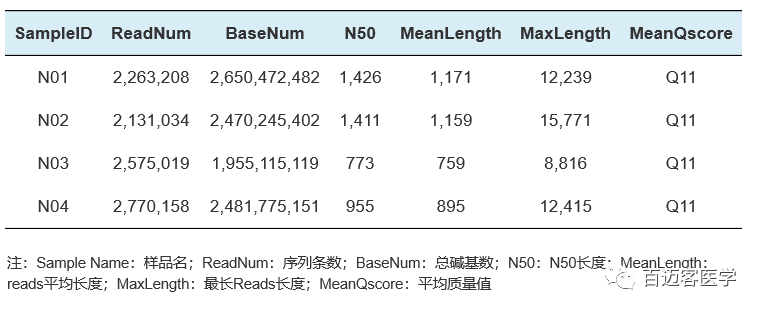
为什么ONT全长转录组测序数据量推荐2G,而二代RNA-seq推荐6G?
ONT全长转录组测序一条reads即代表该转录本表达一次,而二代短reads需要非常多条才能覆盖一个转录本;oxford nanopore公司官方白皮书中数据显示:当相同数量的转录本被覆盖达95%时,ONT所需要的reads数比Illumina约少50倍,所需要碱基数约少7倍。
故而2G ONT数据能达到6G Illumina检测效果;
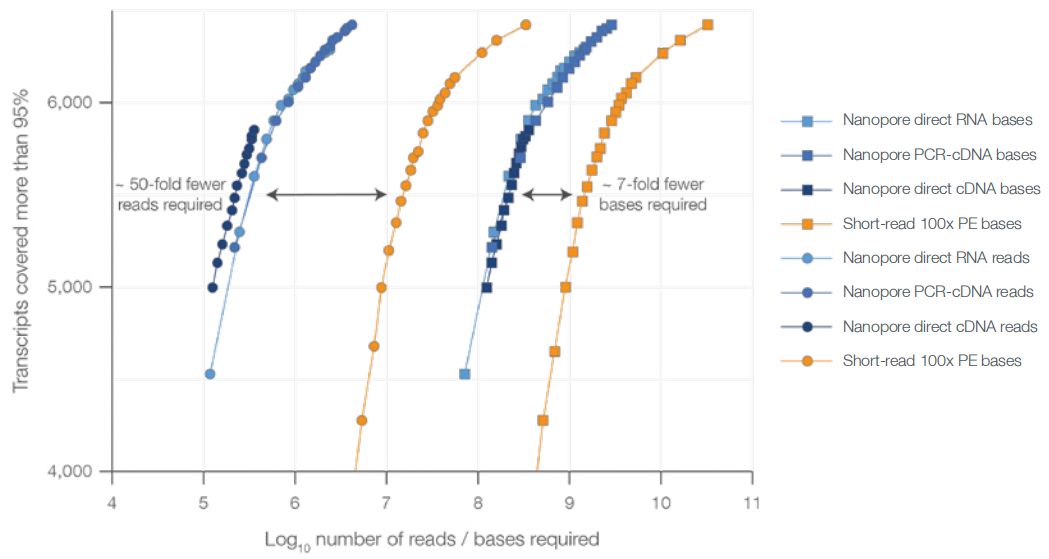
——图片来源于Oxford Nanopore Technologies官方白皮书The value of full-length transcripts without bias。
针对同一样本进行的饱和度分析显示,2G ONT全长除表达量极低的(CPM<1)其他转录本都达到饱和了,和二代Illumina 6G除表达量极低FPKM<1外的基因检测也饱和了,且前者更早趋向饱和;
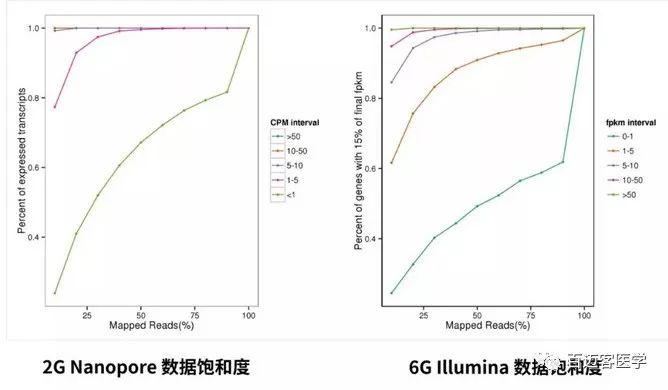
目前已发表的人鼠文献中ONT全长测序的数据量大多也不到2G,比如文献精读|nanopore全长转录组测序揭示B细胞表面受体广泛的转录变异。
ONT全长转录组在可变剪切方向有哪些文章?
应用于慢性淋巴细胞白血病
英文题目:Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns
发表杂志:Nature Communications
发表时间:2020.03
影响因子:11.878
使用Nanopore分别对慢性淋巴细胞白血病(CLL)分离的SF3B1野生型,突变株和正常组B细胞样本进行全长转录组测序。基于Nanopore的全长cDNA测序可以检测转录本全长,通过算法优化,相对于短序列,可以更准确的检测3′末端剪切,内含子保留,分辨生产性异构体和非生产性异构体。该研究证明了Nanopore测序在癌症和可变剪切中的潜在使用价值。
应用于精神疾病
英文题目:Long-read sequencing reveals the complex splicing profile of the psychiatric risk gene CACNA1C in human brain
发表杂志:Mol. Psychiatry
发表时间:2020.03
影响因子:11.973
在人脑中,与精神分裂症相关的基因组区域富集了在神经发育过程中表现出不同异构体使用的基因,本文通过ONT全长转录组技术研究疾病相关的CACNA1C亚型,有可能提供既有效又无外周副作用的新型精神药物。
应用于阿尔茨海默病
英文题目:Deleterious ABCA7 mutations and transcript rescue mechanisms in early onset Alzheimer’s disease
发表杂志:Acta Neuropathol
发表时间:2017.09
影响因子:14.251
阿尔茨海默病(AD)是一种起病隐匿的进行性发展的神经系统退行性疾病。本文研究了ABCA7 PTC突变在一个大型早发性AD对照队列中的患病率和疾病外显性,并用ONT全长转录组检查了其对转录水平的影响。揭示了不同程度的NMD和转录修饰事件,可能影响ABCA7的剂量、疾病的严重程度,并可能为AD的治疗干预创造机会。
应用于多囊肾病
英文题目:Human-Specific Abnormal Alternative Splicing of Wild-Type PKD1 Induces Premature Termination of Polycystin-1
发表杂志:Journal of The American Society of Nephrology
发表时间:2018.10
影响因子:9.274
常染色体显性遗传性多囊肾病的主要形式是由编码多囊蛋白-1(PC1)的基因杂合突变引起的,通过ONT全长转录组测序等方法确认存在多种剪接形式。研究发现,在杂合子个体中,低水平的全长PC1可能会将多囊蛋白信号降低到临界的“成囊”阈值以下。
应用于乳腺癌
英文题目:Nanopore sequencing of full-length BRCA1 mRNA transcripts reveals co-occurrence of known exon skipping events
发表杂志:Breast Cancer Res
发表时间:2017.11
影响因子:4.988
本研究探索了纳米孔测序技术在检测整个BRCA1 mRNA转录本以及对框内和框外剪接事件进行准确分类方面的应用。研究鉴定了32个完整的BRCA1亚型,其中包括18个新的亚型,还发现已知的BRCA1外显子跳跃事件,如Δ(9,10)和Δ21。这些发现对预测剪接转录本的翻译框架具有重要意义,对解释剪接变异体的临床意义也很重要。
应用于肺癌
英文题目:Long read sequencing reveals a novel class of structural aberrations in cancers:identification and characterization of cancerous local amplifications
bioRxiv
本研究中利用ONT全长转录组和ONT重测序技术在肺癌基因组中识别和表征结构畸变,揭示了由局部重复、倒位和微缺失的复杂组合组成的独特结构畸变CLCL,进一步分析并发现,即使在关键的癌症相关基因中,这些突变也发生在体内,这些突变可能阐明了致癌性事件和治疗策略仍然难以捉摸的患者的分子病因。
应用于细胞表面受体
英文题目:Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells
发表杂志:Nature Communications
发表时间:2017.07
影响因子:12.121
短reads RNAseq解析复杂isoform的能力有限,因为它无法测序RNA分子的全长cDNA拷贝。作者研究了使用长读取单分子Oxford Nanopore测序仪的RNAseq是否能够在不牺牲准确的基因表达定量的情况下,鉴定和定量复杂的isoform。在小鼠B1a细胞中鉴定了数千个未注释的转录起始和终止位点,以及数百个可变剪接事件,鉴定了在B1a细胞中表达的数百种基因,这些基因显示出多种复杂的isoform,包括几种B细胞特异性表面受体。本研究表明,可以在单细胞水平上识别和定量复杂的isoform。
案例展示

- 【成功案例】ONT全长转录组联合代谢组解读酰化和非酰化花色苷对Zucker糖尿病肥胖型大鼠肝脏代谢产物和基因表达的影响号外!号外!号外! ONT全长转搭上代谢组带你玩出不一样的高度,且看全长转录组如何推陈出新。百迈客合作单位于2 […]阅读更多
- 【成功案例】借助ONT全长转录组技术解读Colgalt2在非酒精性脂肪肝发病机制中的作用又又一篇高分ONT全长转录组的合作应用文章发表啦!趁着新鲜出炉,小编带大家看看别人的高分文章怎么组装的,又是讲 […]阅读更多
- 【成功案例】–ONT全长转录组分析揭示针刺内观穴改善心肌缺血模型心功能的机制Nanopore全长转录组由于其长读长、低碱基偏好性,同时完成基因水平及转录本水平定量的高性价比的优势,越来越 […]阅读更多
相关解读
- Nanopore全长转录组测序绘制癌细胞系LARP1 isoform表达谱研究背景 La相关蛋白(La-related proteins,LARP)是与癌症相关的RNA结合蛋白(由LA […]阅读更多
转录组相关文章查看所有




Copyright © 2009-2026 北京百迈客生物科技有限公司版权所有 京ICP备10042835号
 京公网安备 11011302003368号-网站地图
京公网安备 11011302003368号-网站地图
京公网安备 11011302003368号-网站地图