
动植物基因组De novo三代测序
专业 准确 完整
产品介绍
动植物基因组De novo测序分析也叫从头测序分析,指不依赖于任何参考序列信息就可对某动植物进行测序分析,使用生物信息学方法进行序列拼接获得某物种的基因组序列图谱,并进行基因组结构注释、功能注释、比较基因组学分析等一系列的后续分析。三代测序技术(以PacBio和Nanopore为代表)具有读长长的特点,自2015年开始在动植物基因组De novo中初露锋芒,已延用至今。该类型测序分析结果可以广泛应用于农林鱼牧医药及海洋等各个方面的研究。

图1 不同测序技术读长,准确性及基因组连续性评估
三代测序技术原理
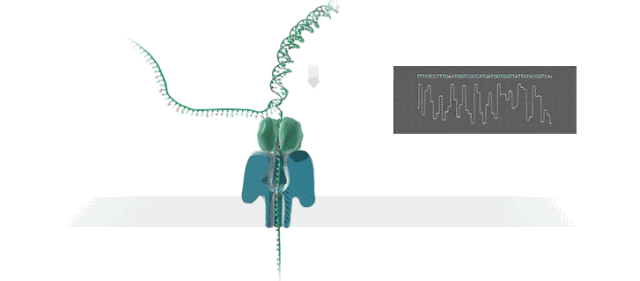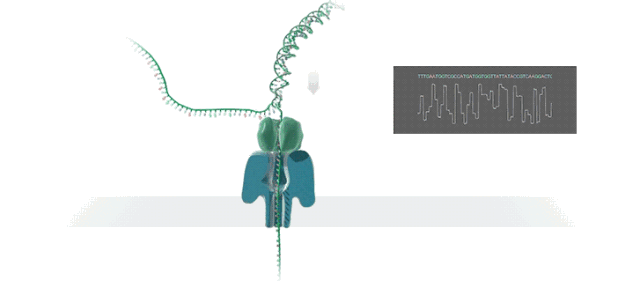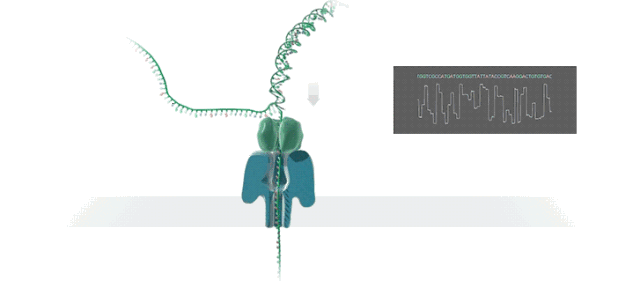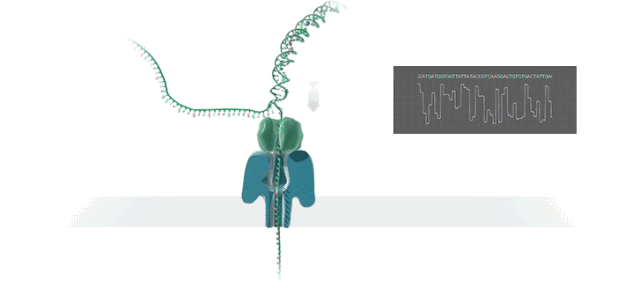
PacBio测序原理
采用边合成边测序的方式,以其中一条DNA链为模板,通过DNA聚合酶合成另外一条链,进一步将荧光信号转变为碱基信号。同时PacBio已升级了CCS测序模式以获得长读长的高保真(HiFi)15 kb reads,由此提升基因组组装的准确性。
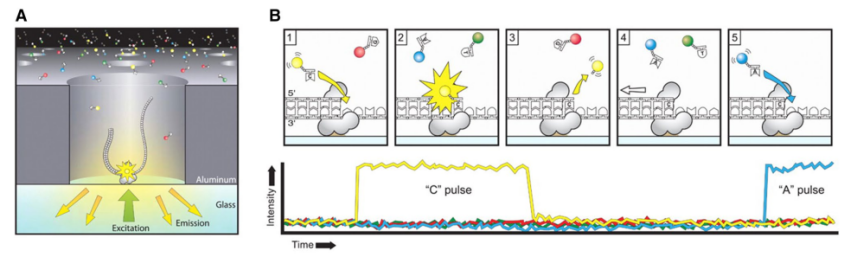
图2 三代PacBio测序原理
Nanopore测序原理
当单链DNA分子穿过纳米孔时,相对于每个核苷酸,都会获得不同的电流信号。记录每个孔的离子电流变化,并基于马尔可夫模型或递归神经网络的方法将其转换为碱基序列。除此之外,Ultra-long reads (ULRs) 是ONT平台的另一重要特征,并具有促进大型基因组组装的潜力。
信息分析内容
| De novo研究 | 研究内容 | |
| 基因组组装 | 多软件组装、组装结果评估 | |
| 基因预测与注释 | 编码基因预测;重复序列注释和转座元件分类;非编码RNA注释;假基因注释等 | |
| Hi-C辅助基因组组装 | 有效数据评估;Contig聚类、排序及定向分析;挂载结果评估 | |
|
生物学问题解析 |
比较基因组学研究 |
基因家族聚类; |
| 系统发育树的构建; | ||
| 基因家族扩张与收缩分析; | ||
| 物种分化时间推算; | ||
| LTR形成时间估算; | ||
| 全基因组复制事件; | ||
| 选择压力分析 | ||
| 特定生物学问题剖析 | 结合组学研究方法,深入对某物种生物学问题进行解析 | |

草莓基因家族聚类分析

薏苡全基因组复制事件分析
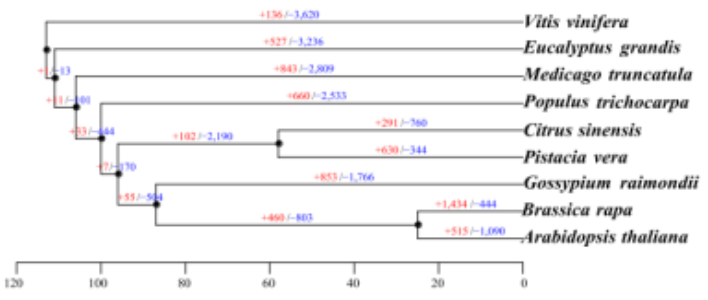
开心果系统进化树与基因家族收缩扩张分析
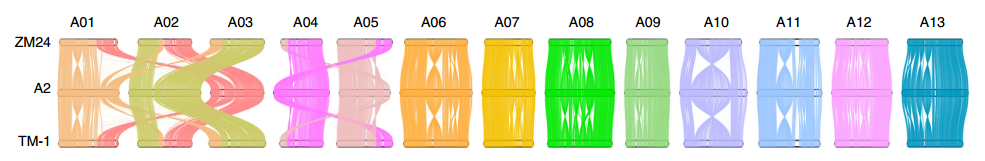
陆地棉亚基因组共线性分析
技术服务流程
-
样品寄送
-
建库测序
-
数据分析
-
出具报告
-
售后答疑
产品优势
公司成立于2009年,深耕基因组测序领域多年,长久以来致力于成为精准的基因组组装专家;
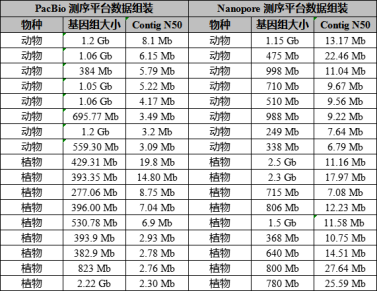
拥有三代测序平台PacBio测序全平台和Nanopore测序全平台,具有丰厚的双平台组装及上万种物种基因组组装经验。
Hi-C染色质构象捕获技术文库有效数据比例高,挂载效率高达99%,多倍体物种研究经验丰富,与三代基因组组装相结合,获得染色体水平基因组的。同时进一步提升基因组组装质量。
项目经验示例
合作文章案例
案例1
以更新的亚洲棉A基因组为基础的243份二倍体棉的重要农艺性状的研究
Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits
期刊:Nature Genetics
影响因子:27.125
发表单位:中国农业科学院棉花研究所、北京百迈客生物科技有限公司等
发表年份:2018年5月
研究背景:
棉花是研究植物多倍化的有价值的资源。亚洲棉(Gossypium arboreum)和草棉(Gossypium herbaceum)的祖先是现代栽培异源四倍体棉花A亚基因组的供体。 本研究中,利用了三代PacBio和Hi-C技术,重新组装了高质量的亚洲棉基因组,分析了243份二倍体棉花种质的群体结构和基因组分化趋势,同时确定了一些有助于棉花皮棉产量遗传改良的候选基因位点。
研究结果:
1、亚洲棉三代基因组组装:
利用三代测序和Hi-C相结合的方法进行亚洲棉基因组组装。共计获得了142.54 Gb ,组装1.71 Gb亚洲棉基因组,Contig N50=1.1 Mb,最长的Contig为12.37 Mb。利用Hi-C技术将组装的1573 Mb的数据定位到13条染色体上,与已经发表的基因组相比,当Hi-C数据比对到更新的基因组后,对角线外的不一致性明显减少(图1 a-b)
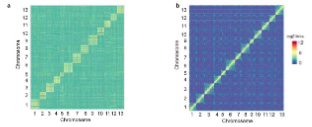
图1 Hi-C数据在两版亚洲棉基因组上的比对
2、二倍体棉花群体遗传进化分析:
对230份亚洲棉和13份草棉重测序,进行基因组比对、系统发育树、群体结构分析、PCA、LD和选择性清除分析得出亚洲棉和草棉(A)与雷蒙德氏棉同时进行了分化;亚洲棉起源于中国南部,随后被引入长江和黄河地区,大多数具有驯化相关特性的种质都经历了地理隔离(图2)。
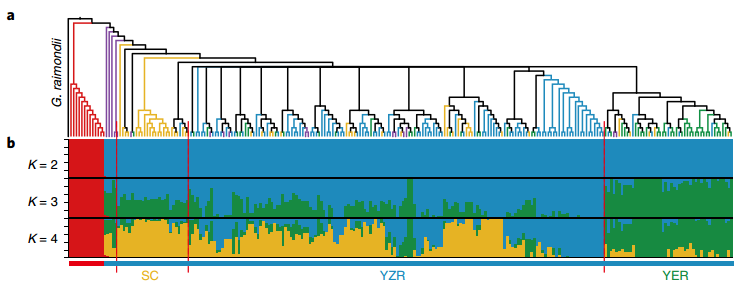
图2 二倍体棉群体进化和群体结构分析
3、亚洲棉的全基因组关联分析(GWAS):
对来自不同环境下的11个重要性状进行全基因组关联分析,鉴定了亚洲棉11个重要农艺性状的98个显著关联位点,GaKASIII的非同义替换(半胱氨酸/精氨酸替换)使得棉籽中的脂肪酸组成(C16:0和C16:1)发生了变化;发现棉花枯萎病抗性与GaGSTF9基因的表达激活相关。选择了亚洲棉种质中的158份有绒毛和57份无绒毛材料进行GWAS关联分析,发现与毛状体和纤维发育有关信息(图3)。
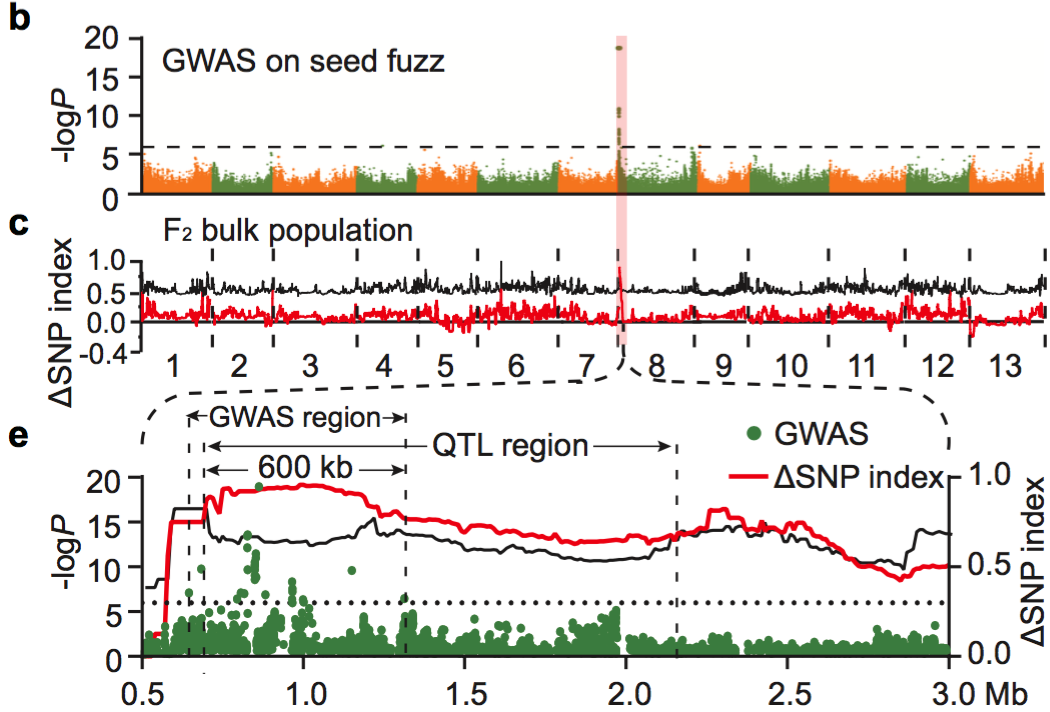
图3 二倍体棉群体进化和群体结构分析
研究结论:
利用三代测序+Hi-C技术完成了亚洲棉基因组的重新组装,将基因组组装指标从72 Kb提升到1.1 Mb,为亚洲棉后续的群体遗传学等相关研究奠定了基础;通过群体遗传进化等相关分析,发现亚洲棉和草棉(A型)与雷蒙德氏棉(D型)同时进行了分化,并证明了亚洲棉起源于中国南部,随后被引入长江和黄河地区;整合GWAS与QTL等分析方法,对亚洲棉脂肪酸含量,抗病性及棉绒生长发育相关基因进行定位,并进行相关功能验证,促进了亚洲棉复杂农艺性状的改良。
案例2、
二倍体、野生和栽培四倍体花生比较基因组分析揭示亚基因组不对称进化和改良
Comparison of Arachis monticola with diploid and cultivated tetraploid genomes reveals asymmetric subgenome evolution and improvement of peanut
期刊:Advanced Science
影响因子:15.804
发表单位:河南农业大学、北京百迈客生物科技有限公司等
发表年份:2019年11月
研究背景:
花生作为我国重要的经济作物,是提供重要的蛋白和油料的基础。花生属一共包括30个二倍体品种,1个异源四倍体野生花生(A. monticola)和1个栽培花生(A. hypogaea)。作为栽培花生农艺性状改良的重要野生资源供体,野生四倍体花生一直是国内外学者的研究热点。研究中对花生属唯一的野生异源四倍体花生Arachis monticola基因组进行了研究,同时对17个野生二倍体花生(AA;BB;EE;KK和CC)与30个野生和栽培四倍体花生进行了重测序分析。
研究结果:
1、野生四倍体花生基因组denovo及与栽培四倍体花生的比较分析:
基于 Illumina、PacBio 、Hi-C和光学图谱数据,组装Arachis monticola(2n = 4x = 40)基因组大小为2.62 Gb ,contigs N50=106.66 Kb,scaffolds N50=124.92 Mb;与栽培四倍体花生A. hypogaea基因组结构变异高度保守,且比野生祖先二倍体更加保守;
2、A、B亚基因组的单系起源和多样性:
对17个二倍体野生种(AA、BB、EE、KK和CC)和30个野生和栽培四倍体花生进行了进化树和PCA分析。结果表明,栽培四倍体花生与野生四倍体花生最接近, A和B亚基因组的单系起源(图1);
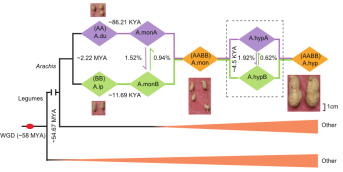
图1 野生和栽培花生的系统进化模型
3、四倍体花生不对称亚基因组进化及表达差异 :
栽培花生和野生花生的亚基因组间的同源序列交换率(HSE)分别为2.46%和2.54%。野生花生中A到B的HSE富集的基因为类黄酮生物合成和昼夜节律途径的基因,暗示不对称HSEs在生物学功能中的作用;
4、SV对荚发育和驯化相关基因表达的影响及抗病基因鉴定 :
对野生四倍体花生和栽培四倍体花生不同发育阶段荚果的SV分析发现SV在荚果发育过程中基因表达的变化上可能起着重要作用;同时在栽培四倍体花生中鉴定到190个SV抗病基因(SV-RGAs),其中32个基因在接种后易感组或抗性组中表现出显著的表达变化(图2)。
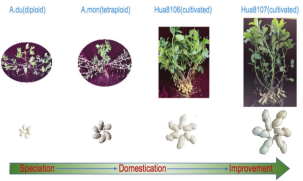
图2 野生花生到栽培品系的豆荚性状驯化
研究结论:
充分注释了高质量野生四倍体花生基因组,揭示了花生亚基因组单系起源和遗传进化模型,表明了野生和栽培四倍体花生亚基因组发生了不对称进化;此外,野生花生中存在的独特等位基因可以改善栽培花生的抗性和荚果大小等形状,为研究多倍体基因组进化、作物驯化和基因组辅助花生生产改良提供独特的价值。



Copyright © 2009-2026 北京百迈客生物科技有限公司版权所有 京ICP备10042835号
 京公网安备 11011302003368号-网站地图
京公网安备 11011302003368号-网站地图
京公网安备 11011302003368号-网站地图