
真核转录组学测序分析
产品介绍
二代真核转录组采用Illumina测序平台,对有参、无参真核生物特定细胞在某一功能状态下转录的所mRNA进行测序。在定量层面,有参可以对基因进行定量分析,无参只能对Unigene(优化的转录本)进行定量分析,并进行下游的差异基因分析和功能注释等;在结构层面,有参可进行可变剪切、SNP分析、基因结构优化、新基因预测。目前已广泛应用于基础研究、临床诊断、药物研发和分子育种等领域。
专业项目方案设计 深度解析数据
转录组可搭配任意其他产品进行多组学的分析,同时为了冲刺高分可选择大样本量方案进行设计。从选材到后续研究内容相关信息的挖掘,整个流程严谨进行,全程跟踪。
-
实验设计
-
RNA提取
-
建库测序
-
数据分析
-
售后答疑
百迈客云,个性化分析全面升级!
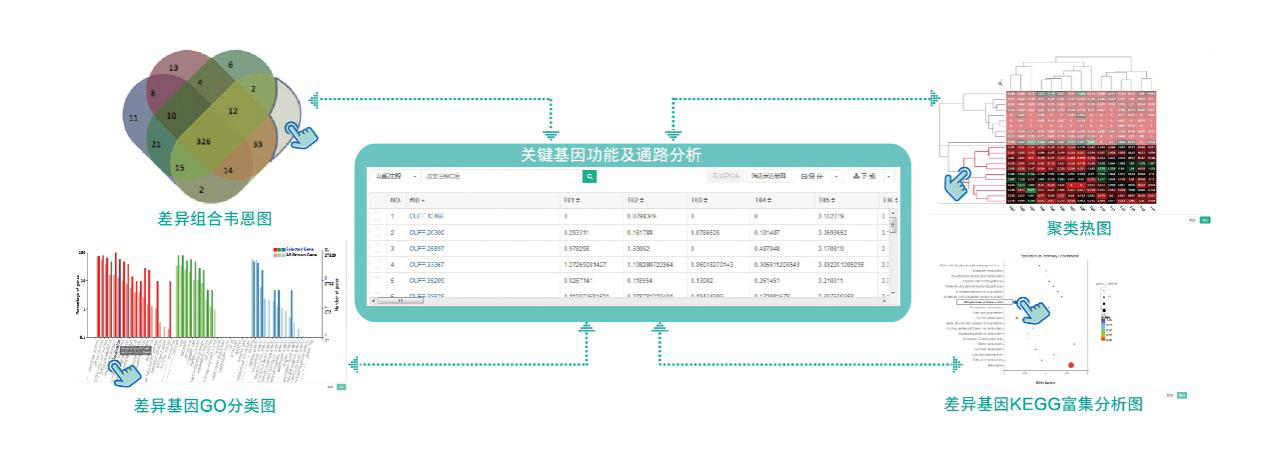
可视化操作、交互性基因深度挖掘,关注哪里“点”哪里。【基本操作】基因功能、基因名称、序列和ID的检索、关键基因功能及通路分析、WGCNA分析等基因功能挖掘。【个性化挖掘】个性化差异分析方案,个性化图表优化、共表达趋势分析、基因结构分析。
结果展示

数据质控
为确保Reads有足够高的质量,将下机原始测序数据(raw reads)去掉含有带接头的、低质量的reads,得到clean reads,保证后续分析的准确性。测序因受测序仪本身、测序试剂、样品等因素影响,存在一定的错误率。碱基测序错误率分布图可以反映测序数据的质量。

参考序列比对
将Clean Reads与参考基因组进行序列比对,获取在参考基因组或基因上的位置信息,定位区域分为Exon(外显子)、Intron(内含子)
和Intergenic(基因间区)。比对到参考基因组上的Reads称为Mapped Reads,Mapped Reads占Clean Reads的百分比,可以评估所选参考基因组组装是否能满足信息分析的需求。

重复相关性评估
生物学重复的相关性不仅可以检验生物学实验操作的可重复性,还可以评估差异表达基因的可靠性和辅助异常样品的筛查。

差异表达基因分析
差异表达基因以火山图、MA图、韦恩图、聚类热图、蛋白互作图等形式呈现,通过火山图(Volcano Plot)可以快速地查看基因在两个(组)样品中表达水平的差异,以及差异的统计学显著性。对于有生物学重复的样本,我们采用DEseq进行样品组间的差异表达分析,获得两个生物学条件之间的差异表达基因集;对于没有生物学重复的样本,使用EBseq进行差异分析。筛选差异基因标准一般为:Fold Change≥2,FDR<0.01。

差异表达基因聚类分析
聚类分析用于判断差异基因在不同实验条件下的表达模式,可通过将表达模式相同或相近的基因聚集成类,从而识别未知基因的功能或已知基因的未知功能,同类基因可能具有相似的功能或共同参与同一代谢过程。

差异表达基因GO分类
差异表达基因GO注释分类统计图,直观的反映出在生物过程(biological process)、细胞组分(cellular component)
和分子功能(molecular function),所有基因和差异基因注释GO term的个数分布。可深入挖掘差异基因的功能及所在的信号通路,筛选关注差异基因注释情况。

差异表达基因蛋白互作网络
STRING收录多个物种预测的和实验验证的蛋白质-蛋白质互作的数据库,包括直接的物理互作和间接的功能相关。结合差异表达分析结果和数据库收录的互作关系对,构建差异表达基因互作网络。

测序数据组装
对于无参转录组,过滤得到的高质量clean reads需通过Trinity软件进行组装得到转录本序列。转录本测序深度除了受测序数据量等影响,还与该转录本的表达丰度有关。为了使各样品中表达丰度较低的转录本组装得更完整,对于同物种的测序样品推荐合并组装可以间接增加测序深度,从而使转录结果更完整,同时也有利于后续的数据分析;而对于不同物种的样品,由于基因组间存在差异,推荐采用分别组装或分开分析。

差异表达基因KEGG通路富集
差异表达基因的KEGG Pathway富集分析,系统分析基因产物在细胞中的代谢途径以及这些基因产物功能,把基因及表达信息作为一个整体的网络进行研究。利用富集因子(Enrichment Factor)分析Pathway的富集程度,并利用超几何检验方法计算富集显著性。
常见问题
在转录组的三个重复测序中,不是所有基因的的可变剪接都会出现3次,有的出现1次或2次,这种情况如何判定该基因是否存在相应的剪接方式?
答:针对每个样品,同一个基因的不同转录本会存在可变剪接,我们只是根据测序的实际数据对可变剪接进行预测,而不是进行验证;如果要判断是否存在相应的剪接方式,需要实验去验证。重复实验存在一定的差异,会导致可变剪接的不同。
转录组结果与基因组比对后,没有相应的注释结果,我们认为是New genes。但是,在NCBI注释后,有些基因的注释结果显示是参考基因组物种,这种基因还能算是new genes吗?
答:我们分析流程中是将测序的Reads比对到参考基因组,然后进行拼接,其中一些reads比对到基因间区并且能拼接出完整的开放阅读框,拼接出来的位于基因间区的这些基因即为新基因。预测得到的新基因才会进行功能注释,所以注释结果与新基因的判断没有关系。
生物学重复样品中某个样品与其他相关性不太好该怎么处理?会不会影响文章发表?
答:为了确保分析结果的准确性,老师通常会设置3个生物学重复,这样就可能出现生物学重复中某个样品相关性不好的情况,影响后续差异分析结果的准确性。通常可将该处理组中相关性不好的样品剔除,再进行差异分析。后期可通过RT-qPCR等试验手段弥补生物学重复的不足,不会影响文章的发表。
如何进行数据挖掘?
答:可从所有基因,差异基因及SNP三个方面进行数据挖掘。所有基因可通过功能注释信息,基因ID,基因名称,序列信息几个方面进行挖掘,同时还可以做表达基因集维恩图,WGCNA等分析。差异基因则可通过维恩图分析不同处理批次几个差异组合共同的差异基因;通常表达量变化趋势一致的基因,可能会有相似的功能,故可通过基因共表达趋势分析来进行差异基因的深入挖掘。SNP则可通过PCA分析,系统进化树,样品间差异SNP筛选及目标区域SNP查询等进行挖掘。以上这些分析均可在我公司云平台免费完成。
KEGG通路注释文件中,K Number Count 指的是什么?
答: K number Count指相关的酶的数目,比如8(6)代表8个基因注释到这个通路,涉及到这个通路的6个酶,某两个基因(或多个)涉及到同一个酶。
测序结果中GO和KEGG富集分析所用的指标分别是KS和Q-value,一般文献用的都是p value<0.05,我想问一下KS和Q-value要分别设置为多少?有没有其他测序的文章用这两个指标。
答:Go富集我们使用的是Blast2GO R包;KEGG是我们根据fisher检验算法自己编写的程序。
KS<0.05,这个值和p-value的意义相同,是TopGO软件包中的一个检验方法。
Q-value<0.01,这个值是对p-value值的一个校正,和FDR概念相似,是fisher检验中的一个检验方法。
测序文章一般不用这两个指标,涉及到算法的文献中才有。
假设有一个基因A,测序结果中有一部分reads可以比对到3‘和5’端,中间的部分没有序列比对上,那么有什么办法可以延伸序列鉴定到中间部分的序列。
答:(1)用实验的方法: 针对5’端和3’端的序列来设计引物,通过PCR实验进行延长和扩增.
(2)生信办法: 将该基因与它的近源物种做同源,如果能找到同源基因,则将该区域的所有read比对到同源基因上,进而来确定中间部分的序列。
项目经验丰富,硕果累累
公司成立多年以来,拥有丰富的项目分析经验,据不完全统计,完成转录组项目10000+,完成样本数200000+;年处理样本数10000+;农学物种涉及粮食作物、果蔬、观赏植物、害虫、家禽牲畜、水产动植物等,医学物种涉及人、鼠,研究方向包括发育调控、环境适应、突变表现、遗传进化、疾病发生发展机制、耐药机制和药物的研发诊断等各种领域。可根据项目需要选择方案,保障结果精准。
成功案例
- 【成功案例】Insm1的SNAG 域调节胰腺内分泌细胞分化并抑制 β细胞转分化为δ细胞2021年5月,百迈客和暨南大学第一附属医院合作的文章“The SNAG Domain of Insm1 Re […]阅读更多

相关文章查看所有
-
转录组+代谢组揭示IbPIF1基因能够提升植株木质素与萜类物质的积累量,进而增强甘薯对茎线虫病的抗性
甘薯茎线虫是一类重要植物寄生线虫,广泛分布于温暖区域;可侵染多种植物,对各类主粮作物的块根造成严重危害。甘薯茎 […]
-
转录组、广靶代谢组测序及分析揭秘水稻最佳收割时期
2026年2月,贵州大学赵全志教授团队在期刊Food Chemistry上发表了一篇题为“Metabolomi […]
-
金龟子绿僵菌通过调节生理、转录组、代谢组及土壤微生物组减轻铅与纳米塑料对水稻的毒性
现有研究表明,农业土壤中铅与纳米塑料复合污染日趋严重,对水稻生长发育、粮食安全及农田生态系统造成双重胁迫,而内 […]
-
转录组学+生理指标+广靶代谢组学揭示小球藻分泌的关键代谢产物可缓解大豆幼苗的盐胁迫
土壤盐渍化是导致大豆产量下降的主要非生物胁迫因素。研究表明,应用小球藻可促进植物生长;然而,其缓解盐胁迫的潜力 […]
-
芒果果皮颜色调控原理被破译,黄酮+关键基因,决定果皮红黄绿
文章题目:Multi-omics analyses unveil the molecular m […]
-
玉米抗病相关文献:DON毒素降42.2%!多组学破解玉米抗病抗毒核心
玉米作为我国主要粮食作物与饲料原料,正面临着禾谷镰孢菌的致命威胁——不仅引发茎基腐病、穗腐病导致大幅减产,还会 […]
-
转录组测序参与揭示CSF1R-PPARα轴在银屑病致病新机制及全身靶向治疗新策略
中山大学中山医学院谈智/周利君教授团队,联合广东省智能科学与技术研究院李昌林研究员及暨南大学附属广东省第二人民 […]
-
LEC2通过植物细胞全能调节剂的表观遗传激活诱导体细胞重编程丨Nature Communications
研究背景 植物体细胞具备较高的发育可塑性,可通过体细胞胚胎发生过程,不经受精作用直接发育形成完整植株,该过程在 […]
-
Nature子刊丨西班牙马德里营养高级研究所发布新型“衰老毒素”,专杀肿瘤中的衰老细胞
2026年1月12日,西班牙马德里营养高级研究所Javier Moral-Sanz和Maria P. Ikon […]
-
福建农林大学揭示寄主昆虫挥发物“反向塑造”寄生线虫觅食行为的新机制研究丨《PNAS》封面文章
2026年2月25日,福建农林大学侯有明院长教授/吴升晏教授团队在国际权威期刊PNAS发表研究成果并入选封面论 […]
-
《JIPB》封面文章:南京农业大学发现改良大豆株型且提高产量和根瘤固氮能力的“一因多效”基因
近日,JIPB在线发表了南京农业大学李艳教授团队题为”Editing a gibberellin […]
-
单细胞与空间转录组整合分析,揭示大麦从分生组织及器官起始细胞命运决定到特定花器官起始过程中的关键转录事件与时空轨迹
2026年1月7日,Nature Plants在线发表了来自德国杜塞尔多夫海因里希·海涅大学的Rüdiger […]
-
佛罗里达大学团队解析玉米重组调控新机制《Nat Commun》
近日,佛罗里达大学的Meixia Zhao教授团队在国际期刊《Nature Communications》上发 […]
-
《Science》:食木性蟑螂和白蚁的营养特化与社会性演化
2026年01月29日,华南师范大学生命科学学院昆虫科学与技术研究所李胜教授团队联合中国科学院分子植物科学卓越 […]
-
《Science Bulletin》印遇龙院士团队研究成果:史氏甲烷短杆菌通过抑制肝脏酮体代谢,导致出生后生长迟缓
近日,岳麓山实验室畜禽品种创制中心印遇龙院士团队在Science Bulletin(IF 21.1)发表研究成 […]
-
Cell | 深渊钩虾基因组揭示其万米超深渊环境的适应性和种群历史
2025年3月,中国科学院深海科学与工程研究所张海滨研究员团队等在深渊钩虾环境适应与种群遗传方面取得新进展,其 […]
-
《Nature》水稻细胞感知病毒侵染并启动广谱抗病毒防御反应的分子机制
农作物生产持续受到由昆虫传播的病毒性疾病的威胁。近年的研究进展揭示了植物抵御病毒病原体所采用的复杂免疫机制。然 […]
-
《Science》ARF3介导的生长素信号对黄瓜性别决定至关重要
近日,国际学术刊物《科学》(Science)在线发表了园艺学院张小兰教授团队在黄瓜生殖发育领域取得的新成果:《 […]
-
J Exp Bot | 扬州大学解析HvERF62基因介导大麦耐渍涝新机制
扬州大学Rugen Xu团队在《Journal of Experimental Botany》杂志发表题为“N […]
-
Mol Plant丨转录组学和群体研究联合助力高粱功能基因组学研究和遗传育种
2025年3月7日,中国科学院遗传与发育生物学研究所谢旗研究员团队联合中国农业大学于菲菲教授团队在Molecu […]




Copyright © 2009-2026 北京百迈客生物科技有限公司版权所有 京ICP备10042835号
 京公网安备 11011302003368号-网站地图
京公网安备 11011302003368号-网站地图
京公网安备 11011302003368号-网站地图