

2025年4月2日,Industrial Crops & Products在线发表了由四川省林草局、四川大学生命科学学院王婧教授课题组联合中南林业科技大学等多家单位合作完成的研究论文,题为” Phylogenomic analysis and SNP fingerprinting construction of Camellia oleifera Abel. germplasm resources using SLAF-seq technology”。该研究利用SLAF-seq技术,对四川省主栽的来自我国南方25个主要油茶品种开展了系统发育和群体基因组学研究,并筛选关键SNP标记构建了DNA指纹图谱。该指纹图谱的构建有望为我国南方尤其是四川地区油茶种质资源保护、开发利用及品种选育提供关键科学依据。
百迈客生物为该研究提供了SLAF-seq测序分析服务。

研究背景
油茶(Camellia oleifera Abel.)是我国重要的木本油料作物之一,以其高含油量与丰富的不饱和脂肪酸而享有“东方橄榄油”的美誉。我国已有超过2000年的油茶栽培历史,并具有约4500万亩的种植面积。油茶不仅具有经济价值,更具重要生态功能。其广泛分布于我国南方多个气候生态带,是研究森林生态适应性与基因资源保护的重要对象。然而,当前油茶品种识别主要依赖形态学方法,容易造成品种混淆、资源管理混乱和知识产权纠纷等一系列问题,难以满足现代林业种质资源保护与良种选育的需求。为推动油茶良种选育,产业升级和响应国家森林四库相关文件的精神,需加快构建高效、廉价且准确的DNA指纹图谱。
研究结果
1.用于SLAF测序的最优限制性内切酶组合
首先,研究共采集25个油茶栽培品种,共100个样本,利用高通量SLAF-seq建库策略进行数据获取。在建库酶切方案筛选方面,团队预先开展酶切模拟试验比较筛选最优组合,Figure 1 显示,RsaI + HaeIII组合在标签分布均匀性、多态性与文库复杂度上表现最佳。共获得362,152个高质量SLAF 标签,其中254,357个为多态性标签,平均GC含量41.30%,平均Q30高达93.44%,显示测序数据具备极高的可靠性。RsaI + HaeIII双酶切组合可实现高效的特异性位点扩增与标签标准化,为后续大规模SNP检测奠定了数据基础。
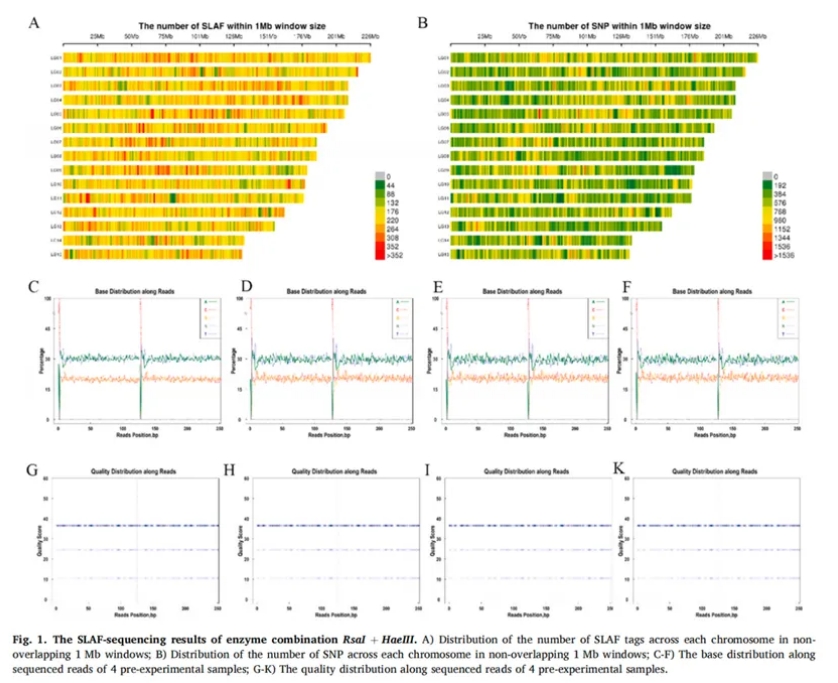
2.基因组变异特征分析
经测序,每个样本平均测序深度为13.95×,共鉴定出470,397个多态性SLAF标签,并初步检测到36,317,329个SNP位点,经过质控过滤后,最终保留了5,685,673个高质量SNP供后续分析。这些SNP广泛分布于油茶基因组的各个染色体区域,具有较强的代表性和应用潜力,为油茶的遗传多样性研究和分子育种提供了宝贵的遗传资源。 Figure 2展示了SNP位点在各染色体上的分布密度、覆盖深度和变异质量参数,为后续的遗传结构分析及品种识别提供了坚实的基础数据支持。
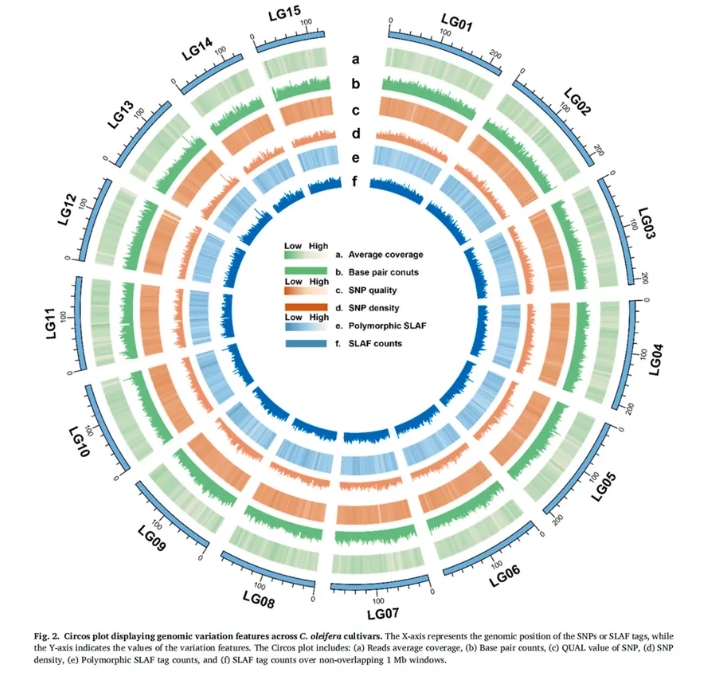
3.各油茶品种的系统发育关系
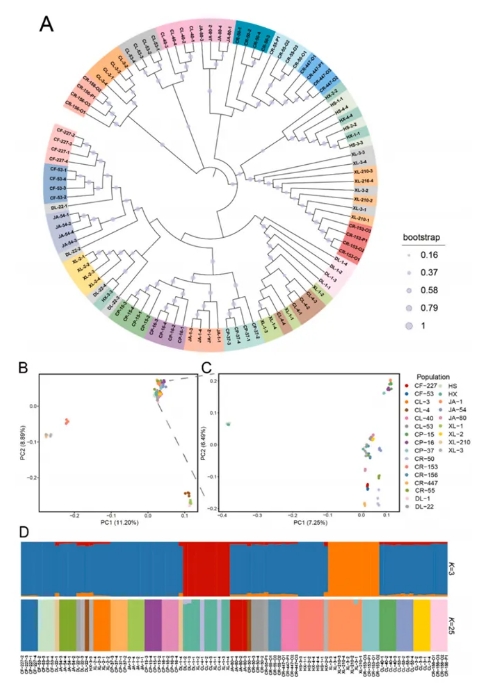
为了系统阐明25个油茶品种之间的亲缘关系与遗传结构,研究结合了邻接法(NJ树)、主成分分析(PCA)与群体结构分析(ADMIXTURE)三种方法,进行了系统发育与群体结构分析(Figure 3)。NJ树的结果显示,大多数样本能够根据其品种进行合理聚类,呈现出明显的系统发育规律。然而,部分品种则呈现出混合的聚类模式,具体包括华硕HS与华鑫HX、湘林XL-3与湘林XL-210、以及湘林XL-1与长林CL-4。而达林DL-22品种的个体聚类较为分散且无序,表现出较为复杂的遗传结构。在PCA分析中,前两个主成分(PC1和PC2)解释了大部分遗传变异(PC1和PC2分别占11.20%和8.89%),并显示出明显的群体结构分布,这与NJ树的聚类结果高度一致。进一步的群体结构分析表明,当K值为3时,分析结果最佳,将品种划分为三大类群:第一类群包括达林DL-1、长林CL-4和湘林XL-1;第二类群包括川荣CR-153、湘林XL-3和湘林XL-210;第三类群则包括其余所有品种。需要注意的是,DL-22品种的个体表现出较高的遗传混杂性,这可能与其基因渐渗、引种或杂交历史相关。这些分析结果为进一步理解油茶品种的遗传多样性、进化关系以及品种改良提供了重要的遗传信息。
4.遗传多样性揭示品种差异
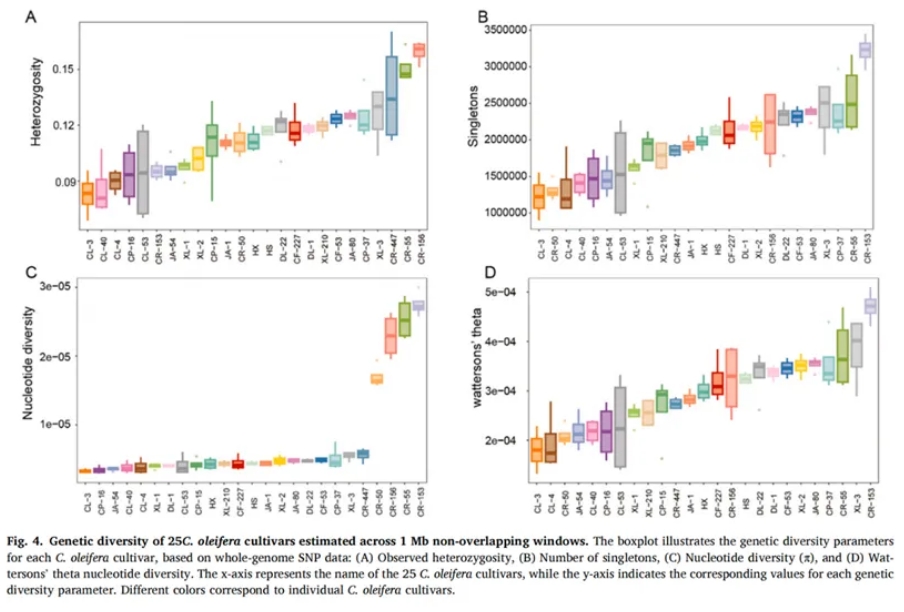
为了全面评估油茶种质资源的遗传多样性,研究统计了包括杂合度(Heterozygosity)、核苷酸多样性指数(π)、单态位点数量(Singleton)以及Watterson’s θ值等关键遗传参数(Figure 4)。结果表明,川荣(CR)系列品种(如CR-153、CR-55)在各项遗传变异指标上均表现出较高水平,尤其是核苷酸多样性指数(π)显著高于其他品种。具体而言,singleton在长林(CL)系列品种(如CL-3、CL-4、CL-40)、翠屏(CP)系列(如CP-16)和江安(JA)系列品种(如JA-54)中较少,而在川荣CR-153和CR-55中则为最多(Figure 4B)。核苷酸多样性(π)值的差异也较为显著,川荣品种CR-50、CR-156、CR-55和CR-153的π值明显高于其他品种,表明川荣系列品种具有较高的遗传多样性,可能为油茶品种改良和分子育种提供重要的遗传资源(Figure 4C)。类似地,Wattersonθ分析进一步揭示了品种间分离位点的变化,川荣CR-153、湘林XL-3和CR-55表现出较高的θ值,与singleton的趋势一致(Figure 4D)。近交系数(F)分析结果与遗传多样性模式一致,显示川荣CR-156、CR-55和CR-447的F值最低,表明其遗传多样性较高,而长林CL系列品种(如CL-3、CL-40、CL-4)的F值较高,反映其遗传多样性较低。总体而言,遗传多样性分析结果表明,长林(CL)系列品种由于长期的人工选育和驯化,遗传背景较为单一,表现出较低的遗传多样性;而川荣系列品种则保持较高的遗传多样性。该研究结果为筛选核心种质资源和制定育种策略提供了有力的遗传依据。
5.遗传分化分析明确亲缘结构
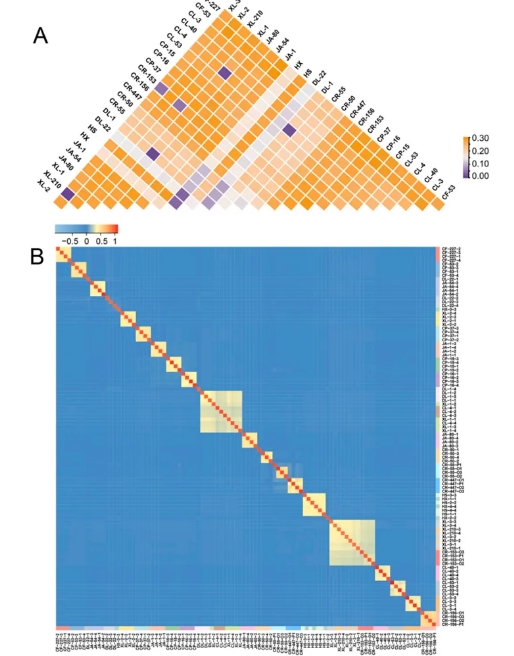
通过计算品种两两之间的Fst值与构建遗传距离矩阵,研究进一步明确了油茶品种间的遗传分化程度和亲缘关系(Figure 5)。Fst值的分析结果显示,长林CL-4、达林DL-1与湘林XL-1三者之间的Fst值极低(0.0106–0.0110),这表明这些品种可能源自相同或相似的遗传背景。类似地,川荣CR-153与湘林XL-3、湘林XL-210之间的Fst值也表现出较高的遗传相似性(0.0036–0.0442),提示这些品种可能具有共同的祖源关系。相比之下,达林DL-22与川荣CR-50(0.0816)、川荣CR-55(0.0870)和达林DL-1(0.0759)之间的Fst值较高,显示出较为显著的遗传分化,反映其与这些品种的亲缘关系较为紧密,但仍保持一定的遗传独特性。其它品种的Fst值范围从0.1013到0.2908,表明这些品种的遗传起源更加多样,可能涉及不同的地理区域或品种间的较大差异(Figure 5A)。研究人员进一步使用GCTA软件计算的G矩阵分析,结果显示大多数油茶品种之间的亲缘关系较为明显,遗传分化较大(Figure 5B)。然而,一些品种群体的亲缘关系值相似,例如XL-1、CL-4与DL-1;CR-153、XL-3与XL-210;华硕HS与华鑫HX,表明这些品种可能源自相同或相近的祖先背景。与此不同,达林DL-22品种在其群体内部未显示出显著的亲缘关系,反映出其群体内存在较高的遗传分化。总体而言,这些分析为理解油茶品种的遗传背景和亲缘结构提供了重要依据,揭示了不同品种之间的亲缘关系及其遗传多样性。
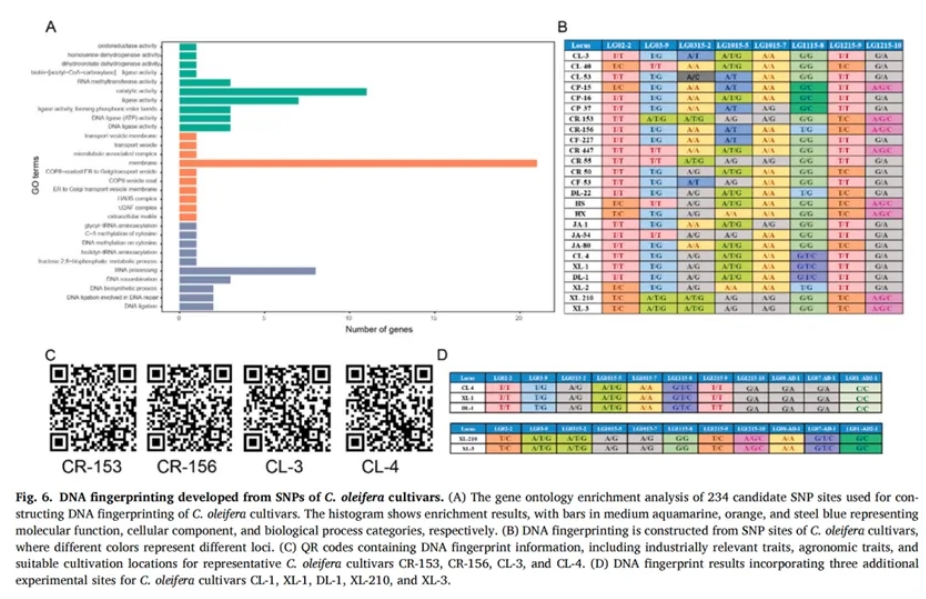
6.构建核心SNP指纹图谱实现品种识别
为建立油茶品种的快速鉴定体系,研究人员进一步构建了25个主栽油茶品种的DNA指纹图谱(Figure 6)。在初步获得的234个候选SNP中,有196个位点被注释为功能明确的编码区域突变,富集于DNA连接、RNA加工、果糖代谢等重要通路(Figure 6A)。为提升区分效率,进一步筛除掉存在多变异干扰或功能不明的位点,最终筛选得到15个核心SNP位点,可有效区分全部25个油茶品种,实现稳定的遗传身份标识。为确保指纹图谱的准确性与应用价值,研究通过Sanger测序对15个候选SNP位点进行了引物验证,最终筛选出8个多态性强、峰型清晰的核心SNP位点用于构建指纹图谱(Figure 6B)。这些位点主要分布于功能基因区,涉及次生代谢、胚胎发育、自噬调控等关键生物过程,如编码α-萜品醇合酶的LG12_G02320和控制胚胎发育与芽形成的FLA8蛋白基因LG03_G02445等,展现出良好的应用潜力。为进一步提升指纹信息的实用性,研究人员还开发了包含8个位点基因型信息的二维码系统,其中涵盖了油脂品质、农艺性状及适宜种植区域等数据,为油茶品种的数字化管理与选育决策提供便捷、高效的技术支持(Figure 6C)。二维码系统为油茶品种的数字化管理与选育决策提供了便捷、高效的技术支持。通过将分子标记信息与油茶生产应用相结合,研究成果不仅实现了分类识别功能,还为分子育种提供了重要参考价值。
总结
综上,该DNA指纹图谱的构建为油茶种质资源的快速鉴定、精准管理及产业化育种提供了高效工具和理论依据。其开发的二维码系统进一步推动了油茶品种管理的智能化和数据化,为我国油茶资源保护、优质品种选育及区域性产业布局提供了科学支持。
内容来源于林木科学评论 侵删




 京公网安备 11011302003368号
京公网安备 11011302003368号