
Hi-C辅助基因组组装
每个基因组都需要一个Hi-C
产品介绍
Hi-C辅助基因组组装是指在已有二代或三代或光学图谱辅助组装的的Draft genome序列和已知染色体数目的前提下,
利用Hi-C测序数据将Draft genome序列进行染色体群组的划分,
并确定各序列在染色体上的顺序和方向,使基因组组装组装水平提升到染色体水平的技术。

不借助遗传图将基因组挂载到染色体水平。每一个基因组都需要一个Hi-C
HI-C特点
分析内容

服务流程
-
样品寄送
-
建库测序
-
数据分析
-
出具报告
-
售后答疑
产品优势
百迈客生物成立于2009年,深耕基因组测序领域多年之久
是目前国内为数不多的基因组测序服务公司
其拥有二代测序仪,三代测序仪(RS II 和sequel),光学图谱Irys,Hi-C组装技术等
拥有自主研发的基因组测序和分析技术,目前已经获得30多项发明专利,超过150多项核心软件著作权
三代+光学+HIC拯救复杂基因组—-大麦基因组Nature篇
新鲜出炉的消息,新版大麦基因组又发nature啦!
大麦基因组到底经历了怎样的困局?到底是什么解救了大麦基因组?后续大麦基因组还会发Nature吗?重复序列比例过高如何解决?新技术的到来更新基因组的必要性到底如何?
大麦基因组的困局:
大麦作为重要的经济作物,其在农业上的重要性毋需本编过多描述。大麦基因组破解工作本处于第一梯队,为何初版基因组在2012年才发布呢?原因就是大麦基因组的属于高重复的复杂基因组,通过当前技术是无法很好解决的。虽然只有7条染色体,但是基因组的重复序列比例高达84%,同时基因组大小在5.1 Gb,相比于人,水稻等简单基因组,技术上存在很大的难度。
和人类基因计划一样,通过集齐全世界科学家的努力,构建了大量的BAC文库,得到了物理图,同时基于遗传图谱,得到了初版基因组。虽然通过综合各种技术,得到的了基因组序列在4Gb 左右,但其可靠性,准确性难以保障。就拿二代数据来说,当时只组装出了1.9 Gb contig的序列,指标更是无从说起。虽然全世界科学家的努力不可否认也不容质疑,但现在看来,初版大麦基因组给人的感觉只能是有胜于无!
到底是什么解救了大麦基因组?
废话不多说,看看人家的组装结果(表1)。
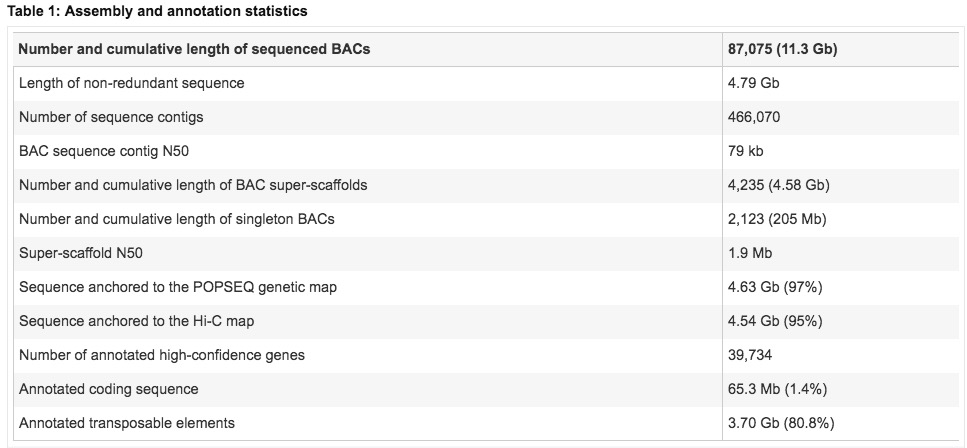
文章中使用的技术手段包括BAC+Illumina+BioNano+HiC+Genetic Map,得到了4.79 Gb基因组序列,最终利用HIC和遗传图分别将95%和97%的序列挂到了染色体的水平。相比于初版基因组,组装水平高了不只是一大截啊,这就是技术上的胜利!话说,基于此版基因组,预测出的基因编码区至占到了整个基因组的1.4%,而转座原件(重复序列的一个大类)却占到了整个基因组的80.8%。所以说,大麦基因组的难度的确大啊!
请看文章中描述的组装技术路线:
构建87085个BAC,利用Hiseq 进行PE及MP文库测序得到4.5 Tb二代数据,之后将每个BAC的测序数据分别进行组装;
通过物理图谱将BAC间的关系确定;
利用遗传图+光学图谱,通过组装好的BAC序列构建Superscaffold;
利用群体遗传图(POPSEQ)进行Superscaffold分组(97%分组);
利用HiC进行Superscafold排序及定向(95%挂载);
基因组评估+基因预测+后续分析。
文章中有哪些意思的点?(文章中都做了啥分析?)
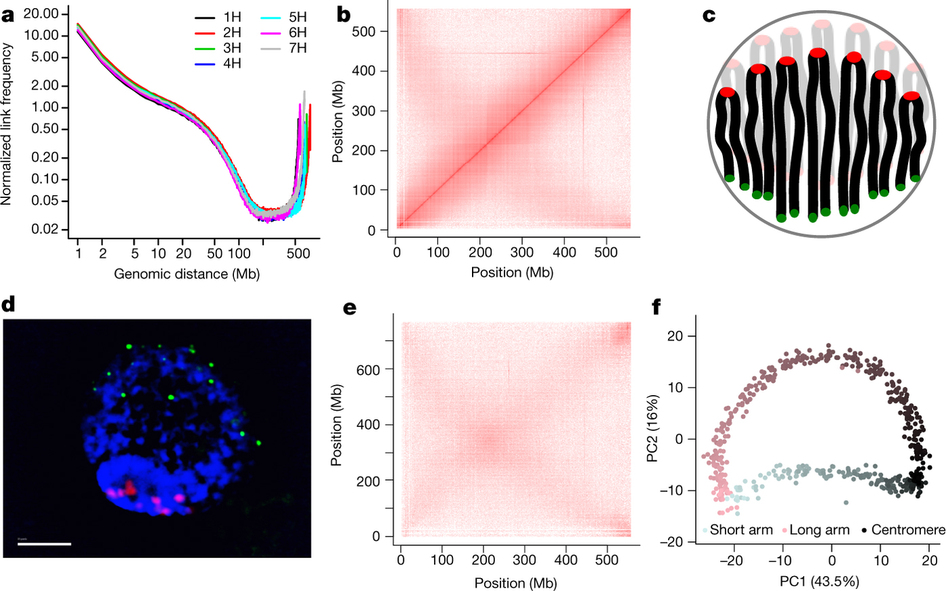
1. 染色体间外大小臂之间的交互
通过HIC热图作者发现无论是染色体内部还是染色体间的长短臂之间都存在较强的交互信号。按照HIC的原理来说,染色体上空间作用越强则实际DNA间的物理距离越近,染色体大小臂及不同染色体间的相互作用应该是极弱的。为了找出原因,作者通过对大麦叶核间期的细胞进行着丝粒及端粒荧光杂交,发现所有染色体的端粒和着丝粒在空间上的位置都纯在极性,且排列方式也极其相似,不同染色体间的大小臂其实在空间上距离很近,因此确实存在染色体内外大小臂之间大量的交互作用的可能。
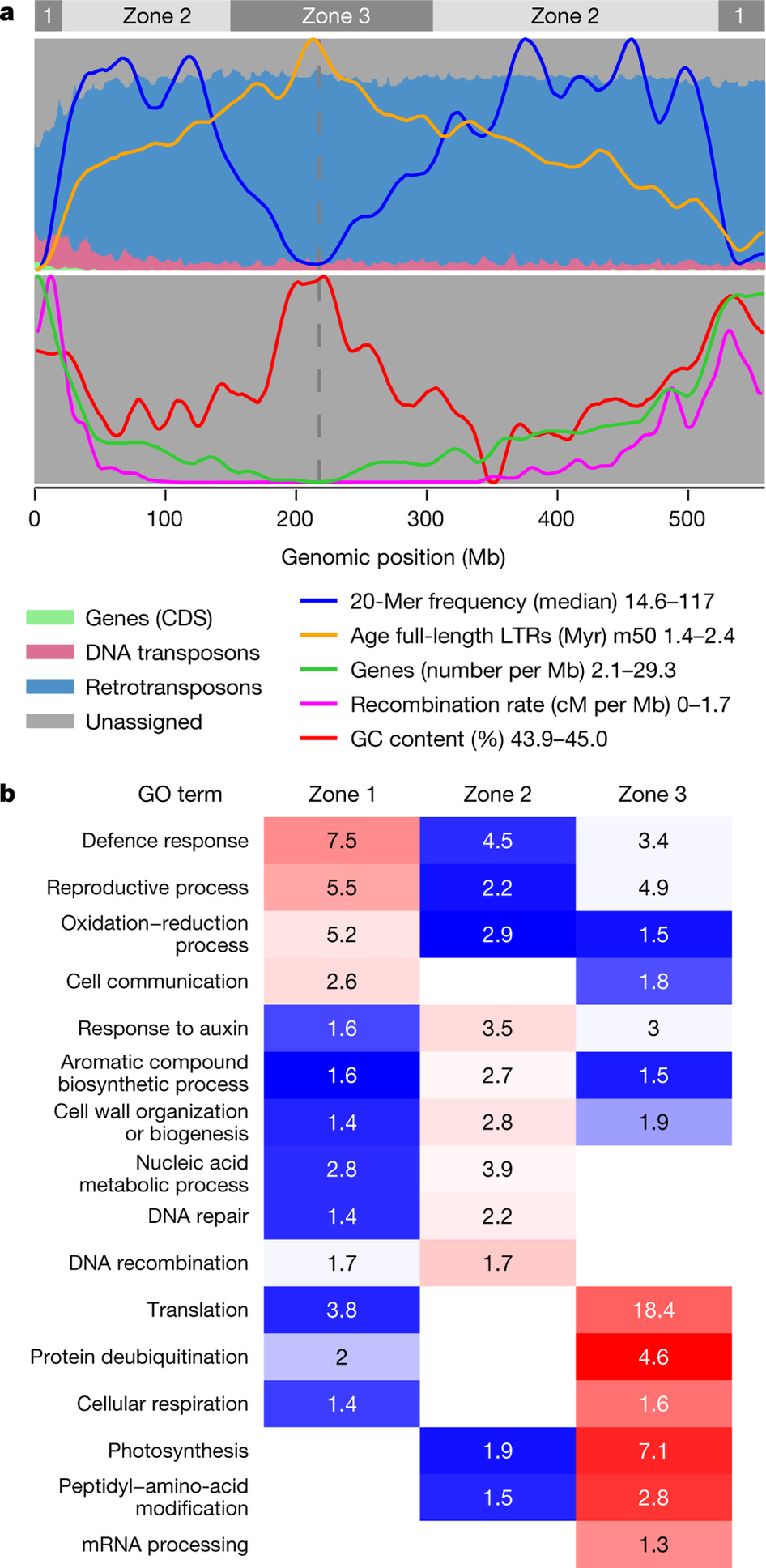
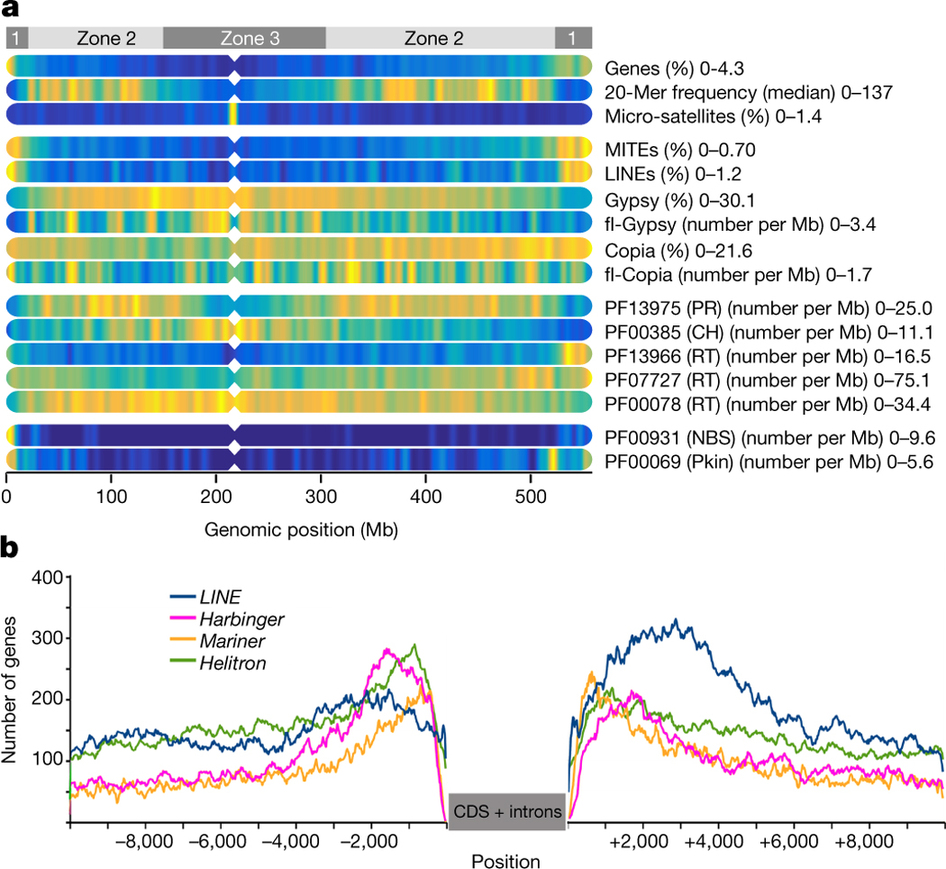
染色体上重复序列及基因密度
利用染色体位置信息,通过对20-mer频率将染色体进行划分成了三种区域,每种区域上在基因密度,重组率,LTR插入时间以及GC含量上都存在一定的规律。
基因家族分析
通过对大麦基因组进行基因家族收缩扩张分析发现,收缩扩张的家族中最显著的部分都与植物防御及抗病相关。另外,作者对麦芽品质相关的amy家族及糖代谢相关的SWEET家族进行了亚家族分类,多倍化及表达模式相关的分析。
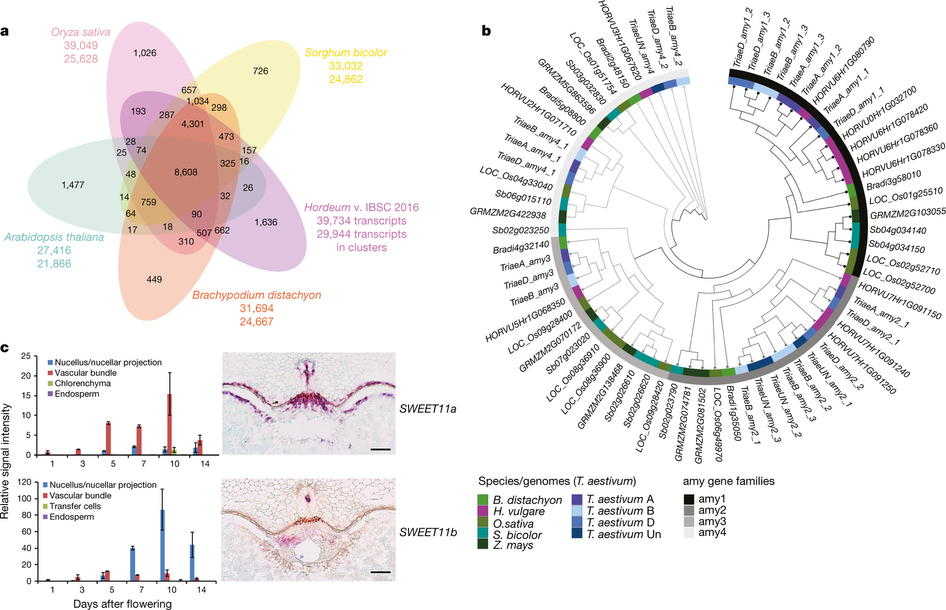
遗传多样性及单体型分析
基因组在分子遗传育种中具有极其重要的作用,本文中作者对来自欧洲的冬季及春季小麦两个群体进行了遗传多样性及单体型相关分析。最终发现,这两个群体在不同的染色体位置上的多样性程度及连锁强度都存在不同特点。如果没有一个好的基因组,很难全面了解群体间的变异情况,会给功能育种上带来困难。
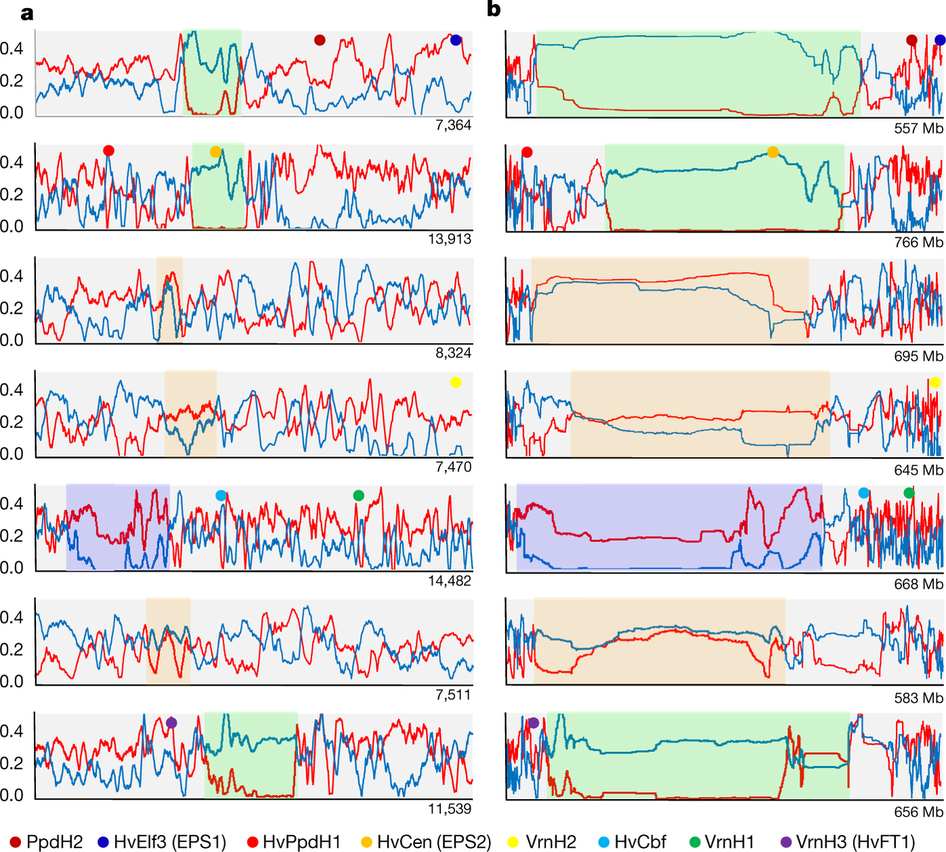
大麦基因组还能发Nature吗?重复序列比例过高如何解决?
虽然此版基因组已经发表,但是本编觉得就目前的技术而言,大麦基因组还是有很大的提升空间。有咩有发现,此版大麦基因组没有使用当前主流基因组所使用的三代测序技术?虽然此版本基因组相较于第一版基因组提升较大,但是基因组装的过于零碎仍旧是事实。毕竟此版基因组的contigN50才79Kb,而super scaffold N50也才1.9Mb。一旦过于零碎,肯定会导致许多基因无法被预测出,这将对后续基因组的功能解读及研究增加困难。目前,三代测序技术在基因组完整性上能够有很好的发挥,同时在基因组结构变异上也能够有所保障。针对大麦基因组,已经有了如此多的数据,本编认为,如果后续如果再加入纯三代测序数据,contigN50达到Mb级别是极其轻松的!在这里可以和大家透露下,本编最近接触到另一个高重复的物种(预测出的重复序列比例高达84%),通过纯三代+HiC组装,在组装指标及完整性上都秒杀了此版本的大麦。所以大麦还会不会发Nature,大家都应该明白了!
A chromosome conformation capture ordered sequence of the barley genome
什么是Hi-C技术?
Hi-C技术是染色质构象捕获技术( Chromosome conformation capture )与高通量测序( High-throughput sequencing )结合衍生的一种技术。主要是利用全基因组范围内整个染色质DNA在空间位置上的关系,对染色质内全部DNA相互作用模式进行捕获,结合生物信息学方法,来获得染色体水平的基因组序列并得到染色质三维结构信息。此外还可以并与Chip-seq、转录组数据联合分析,从基因调控网络和表观遗传网络来阐述生物体性状形成的相关机制。
Hi-C在辅助基因组组装时有什么作用?
- Hi-C最主要的作用是将零散的基因组序列锚定到染色体上(这一点类似遗传图谱);
- 还可以对组装的基因组进行纠错处理;
- 在某种程度上进一步提升Contig N50.
Hi-C技术与遗传图谱的差异?
- Hi-C应用单个个体就可以完成染色体构建;
- Hi-C挂载染色体效率高达90%以上;
- 但Hi-C技术不能进行QTL定位。
Hi-C技术的样品要求?
- 植物样品要选择活体幼苗;
- 动物样品先用全血;
- 其他样品请咨询百迈客。



Copyright © 2009-2026 北京百迈客生物科技有限公司版权所有 京ICP备10042835号
 京公网安备 11011302003368号-网站地图
京公网安备 11011302003368号-网站地图
京公网安备 11011302003368号-网站地图