
近日,国际知名综合期刊PNAS 在线发表了复旦大学、苏州大学等单位联合完成的重磅研究成果,题为“The SWI/SNF chromatin-remodeling subunit DPF2 regulates macrophage inflammation in intestinal injury via CACNA1D-mediated MAPK pathway。”

该研究突破性地揭示了SWI/SNF染色质重塑复合物亚基DPF2,是调控巨噬细胞炎症极化与肠道损伤进展的关键分子,阐明了DPF2通过CACNA1D-钙信号-MAPK 轴驱动巨噬细胞促炎活化的全新机制,为肠道辐射损伤与炎症性肠病的靶向治疗开辟了全新方向。百迈客生物为该研究提供了百创空间转录组BMKMANU S3000测序服务!
研究背景
腹部盆腔肿瘤放疗引发的肠道辐射损伤(RIII)、以及炎症性肠病(IBD),是临床高发的肠道炎症性疾病,常伴随肠道黏膜屏障破坏、持续炎症浸润与上皮再生障碍,目前仍缺乏安全有效的靶向治疗手段。巨噬细胞作为肠道固有免疫的核心细胞,其炎症极化失衡是驱动肠道慢性炎症与组织损伤的关键枢纽,然而巨噬细胞亚群的功能异质性、原位空间分布特征及其在肠道损伤微环境中的调控机制,仍未被完全阐明。
既往针对肠道炎症的研究多采用bulk测序或单细胞测序,无法还原免疫细胞在肠道黏膜复杂结构中的原位空间定位与微环境互作特征,难以解析 “特定细胞亚群如何调控全肠道炎症应答” 的核心科学问题,成为该领域长期难以突破的技术瓶颈。
研究结果
DPF2缺失显著缓解肠道辐射损伤与结肠炎病理表型
为明确DPF2在肠道炎症损伤中的核心功能,该研究首先构建了Dpf2全身杂合敲除小鼠(Dpf2+/-),通过基因鉴定、RT-qPCR与Western blot验证了Dpf2的敲除效率,证实杂合敲除未影响小鼠肝脏、皮肤、肠道等重要器官的正常生理结构与稳态,为后续体内功能实验奠定了基础。通过全身辐照(WBI)肠道损伤模型:对野生型(WT, Dpf2+/+)与Dpf2+/-小鼠进行12 Gy X射线全身辐照,辐照后3天取材分析。
结果显示,Dpf2+/-小鼠的肠道病理损伤显著缓解,肠道绒毛高度与完整性显著优于WT对照组,肠道上皮细胞凋亡水平显著降低(Fig 1A-D);同时,RT-qPCR结果显示,Dpf2+/-小鼠肠道组织中Il1b、Il6、Il18、Ifng、Ccl2等促炎细胞因子与趋化因子的mRNA表达水平较WT组显著下调(Fig 1E-H)。上述结果首次证实,DPF2是肠道炎症损伤进展的关键正向调控因子,其缺失可显著抑制肠道炎症应答,缓解辐射与化学损伤引发的肠道病理损伤。
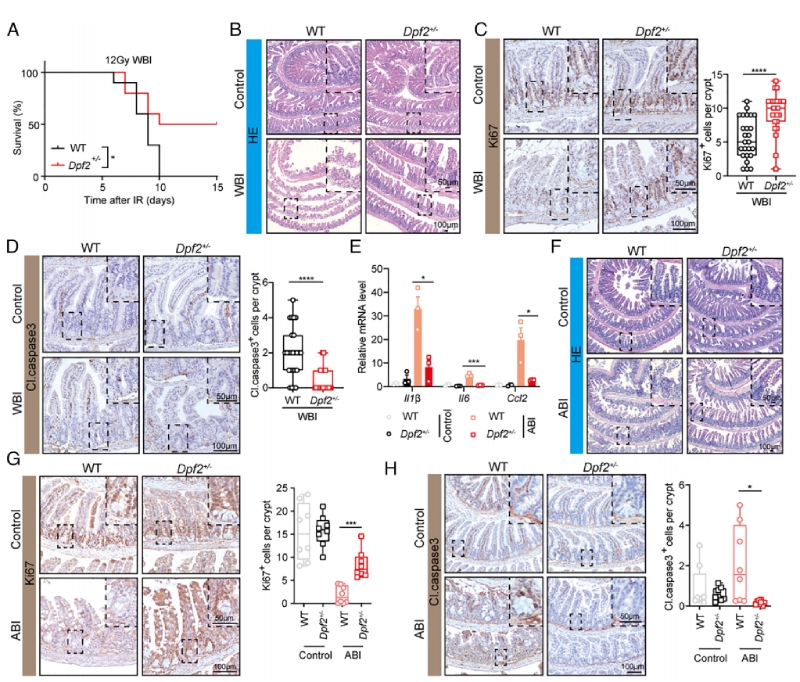
图1
巨噬细胞是DPF2调控肠道辐射损伤的核心功能细胞
为明确DPF2发挥肠道炎症调控作用的核心细胞类型,该研究通过细胞特异性敲除与骨髓移植,锁定DPF2发挥作用的关键细胞类型:该研究分别构建了Lgr5+肠道干细胞特异性Dpf2敲除小鼠(Lgr5-CreERT2; Dpf2fl/fl, Dpf2ΔLgr5)、肠道上皮细胞特异性Dpf2敲除小鼠(Villin-CreERT2; Dpf2fl/fl, Dpf2ΔVil)。10 Gy腹部辐照后的实验结果显示,两种细胞特异性敲除小鼠的肠道促炎因子表达、肠道结构损伤程度、杯状细胞数量、上皮增殖与凋亡水平,均与WT对照组无显著差异(Fig2A-D),证实肠道干细胞、肠道上皮细胞中的DPF2不参与肠道辐射损伤的调控,完全排除了非免疫细胞的功能贡献。
骨髓移植实验锁定造血免疫细胞:该研究设计了经典的骨髓移植实验,将WT或 Dpf2+/-小鼠的骨髓造血干细胞,通过尾静脉移植到经11Gy致死剂量辐照的WT受体小鼠中,重建受体小鼠的造血免疫系统,1个月后对受体小鼠进行10 Gy腹部辐照。结果显示,接受Dpf2+/-骨髓移植的受体小鼠,生存率显著高于接受WT骨髓移植的对照组;辐照后3天,接受WT骨髓移植的小鼠肠道绒毛结构完全丢失、隐窝崩解、炎症细胞广泛浸润,而接受Dpf2+/-骨髓移植的小鼠肠道绒毛结构相对完整、隐窝呈增殖态、黏膜层炎症细胞浸润极少;同时,受体小鼠肠道中Il1β、Il6、Ccl2、Cxcl1等促炎因子的表达水平显著降低,肠道上皮增殖能力显著增强,凋亡水平显著降低(Fig 2E-J)。
在此基础上,进一步构建巨噬细胞特异性敲除(Dpf2ΔCsf1r),复现全身敲除的肠道保护表型,证明DPF2通过调控巨噬细胞,而非上皮或干细胞,介导肠道辐射损伤进程。
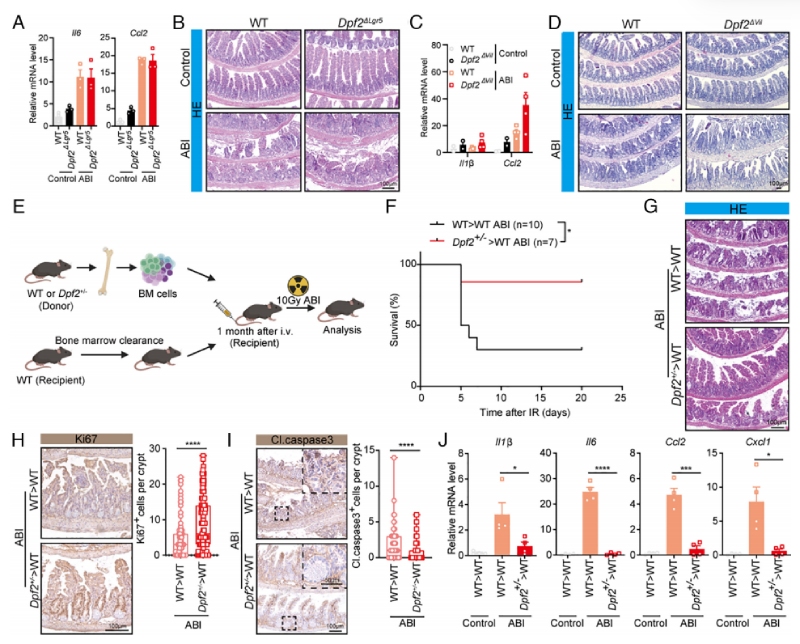
图2
DPF2缺失重塑巨噬细胞极化抑制促炎表型
基于骨髓移植实验的结果,该研究进一步锁定了巨噬细胞这一核心功能细胞,首先构建了H11示踪小鼠与Dpf2ΔUBC小鼠的杂交品系,将其骨髓细胞移植到WT受体小鼠中,经他莫昔芬诱导实现免疫细胞特异性Dpf2敲除后,对小鼠进行10 Gy腹部辐照,随后通过流式分选富集肠道免疫细胞,完成单细胞RNA测序(scRNA-seq)。实验最终捕获到19713个高质量免疫细胞,经质控与无监督聚类,注释得到11种主要免疫细胞类型,包括B细胞、巨噬细胞、T细胞、NK细胞、树突状细胞等(Fig 3A-B)。对巨噬细胞进行亚群细分,得到7个独立的巨噬细胞亚群,其中WT辐照组(WTIR)的巨噬细胞以Cluster 1为主,该亚群显著富集IL-1介导的急性炎症应答通路,高表达M1型促炎标志物Nos2;而Dpf2敲除辐照组(KOIR)的巨噬细胞以Cluster 2为主,该亚群显著富集组织重塑相关通路,高表达M2型抗炎标志物Ccl22(Fig 3C-D)。
为进一步验证scRNA-seq的结果,作者构建了巨噬细胞特异性Dpf2敲除小鼠(Csf1r-iCre; Dpf2fl/fl, Dpf2ΔCsf1r),该模型可在单核/巨噬细胞中实现Dpf2的特异性敲除。流式细胞术结果显示,LPS诱导M1极化、IL-4诱导M2极化后,Dpf2敲除显著降低了M1型巨噬细胞的比例,同时显著提升了M2型巨噬细胞的比例(Fig 3E-F)。10 Gy腹部辐照后的体内功能实验结果显示,仅在巨噬细胞中特异性敲除Dpf2,即可完全复现全身敲除的肠道保护表型:Dpf2ΔCsf1r小鼠的肠道黏膜损伤显著减轻,Il1β、Il6等促炎因子表达显著下调,肠道绒毛结构完整性、杯状细胞数量、上皮增殖能力显著提升,上皮凋亡水平显著降低(Fig 3G-J)。
上述结果最终证实,巨噬细胞是DPF2调控肠道炎症损伤的非冗余核心功能细胞群,DPF2缺失可通过调控巨噬细胞的极化表型,抑制其促炎活化,发挥显著的肠道保护作用。
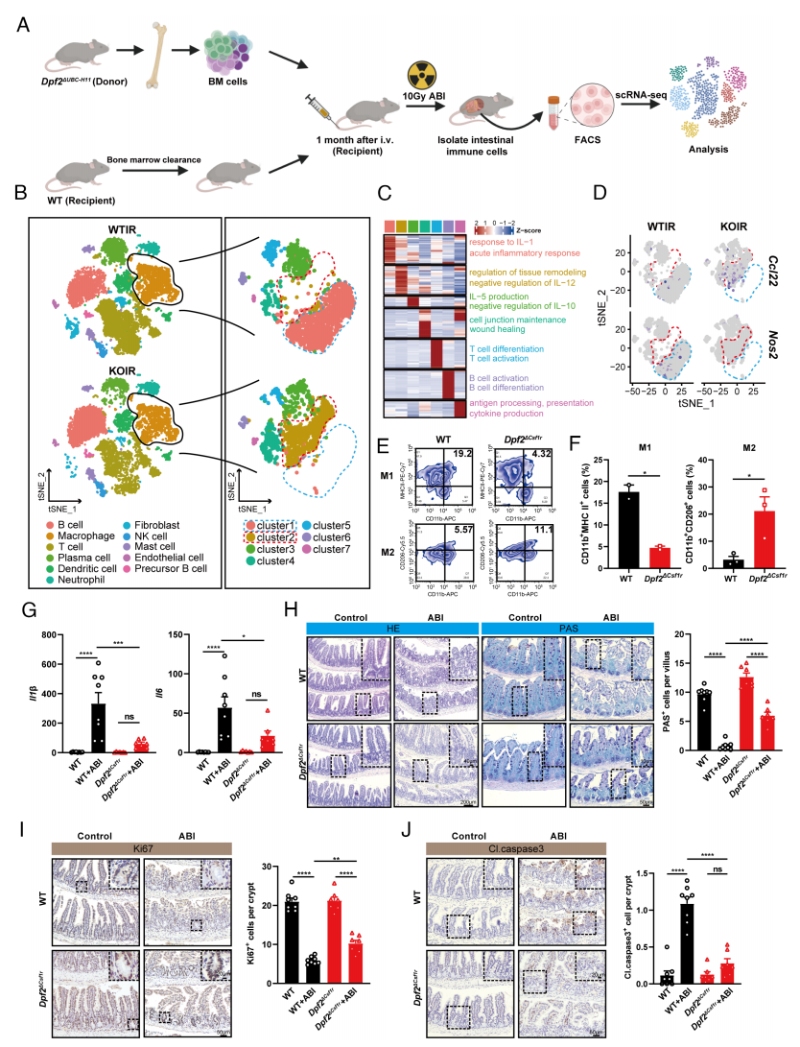
图3
百创S3000空间转录组原位解析DPF2调控肠道炎症微环境的空间分子特征
为在原位空间层面破解DPF2缺失后,巨噬细胞在肠道黏膜精细结构中的空间分布、转录特征与微环境互作规律,该研究采用百创 S3000空间转录组测序平台,对WTIR组与KOIR组小鼠肠道组织进行了高分辨率空间转录组分析。基于百创S3000的超高分辨率数据,精准完成了肠道组织的空间细胞分群,注释得到15种肠道主要细胞类型,包括肠细胞、杯状细胞、潘氏细胞、干细胞、TA 细胞、tuft 细胞、肠内分泌细胞、巨噬细胞、成纤维细胞、内皮细胞等,并完整还原了各类细胞在肠道绒毛-隐窝轴上的生理空间分布特征-干细胞、潘氏细胞特异性定位于隐窝底部,肠细胞沿绒毛从底部到顶端呈梯度分布,巨噬细胞主要富集于黏膜固有层,与肠道生理结构完全匹配;空间分群显示,Dpf2敲除组肠细胞比例更高、损伤细胞更少,干细胞比例更高;KOIR组肠道吸收功能更强,Il1b、Ccl2、Cxcl1等促炎因子空间信号显著降低(Fig 4A-F)。WTIR组巨噬细胞大量募集到受损上皮区域;KOIR 组巨噬细胞维持在固有层稳态位置(Fig 4G)。Dpf2缺失使巨噬细胞分化轨迹趋于单一稳态,促炎通路显著下调,抗炎通路上升(Fig 4H-M)。
空间层面结果显示,KOIR组的巨噬细胞中,M1型促炎通路显著下调,M2型抗炎与组织修复通路显著激活,与scRNA-seq的分析结果一致,实现了单细胞转录组特征的原位空间验证。
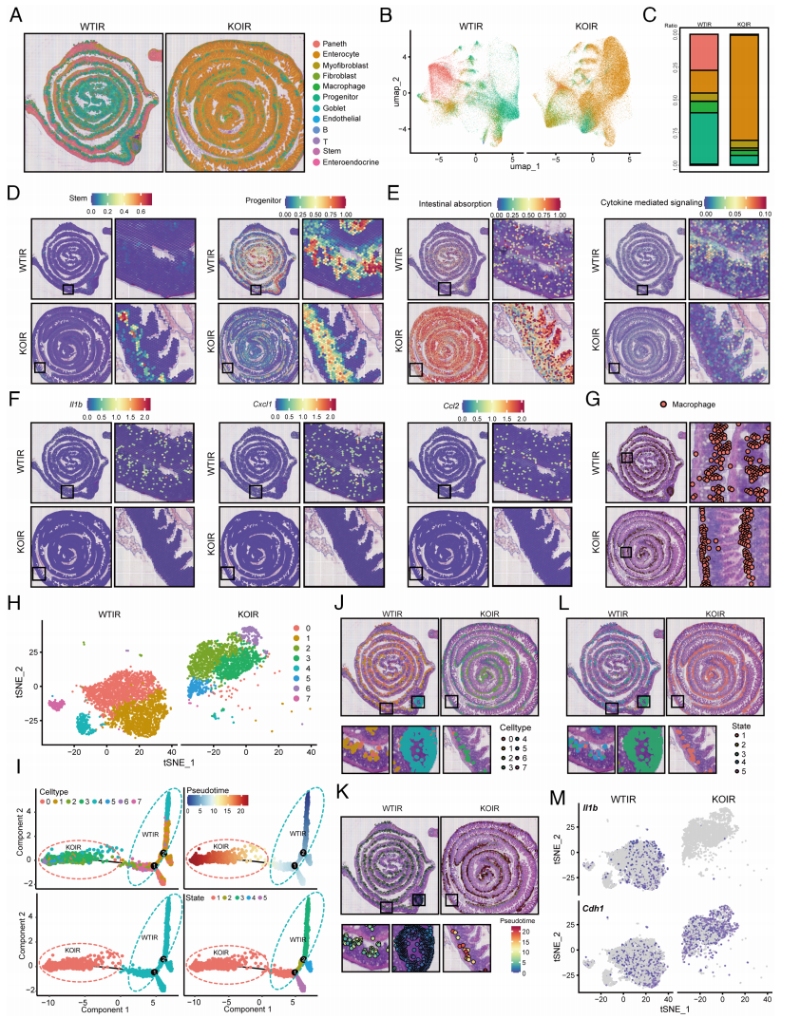
图4
DPF2通过表观遗传调控直接激活CACNA1D转录介导钙信号-MAPK通路驱动巨噬细胞促炎极化
为明确DPF2调控巨噬细胞炎症极化的分子机制,该研究首先对scRNA-seq数据中的巨噬细胞亚群进行差异表达分析,发现CACNA1D是DPF2缺失后巨噬细胞中下调最显著的基因,且在巨噬细胞中呈特异性高表达(Fig 5A-B)。CACNA1D基因编码L型钙通道Cav1.3的α1D亚基,是电压门控钙通道家族的核心成员,也是调控细胞外Ca²+内流、胞质钙信号稳态的关键分子,而钙信号是巨噬细胞活化与炎症因子产生的核心上游调控信号。RT-qPCR验证结果显示,巨噬细胞特异性DPF2敲除后,肠道免疫细胞中CACNA1D的mRNA表达水平显著下调(Fig 5C)。
为验证DPF2对CACNA1D的直接表观遗传调控作用,通过ATAC-seq结果显示,DPF2敲除后,巨噬细胞中CACNA1D基因区域的染色质可及性显著降低(Fig 5D);巨噬细胞ATAC-seq的通路富集分析结果显示,DPF2敲除后,钙信号通路、细胞因子-细胞因子受体互作、MAPK信号通路均发生显著富集,进一步验证了该调控轴的核心地位(Fig 5E)。DPF2缺失降低巨噬细胞胞内Ca²+水平,抑制p38/p44/42 MAPK磷酸化(Fig 5F-G)。敲低CACNA1D上调抗炎因子;TGF-β中和抗体可逆转DPF2敲除的肠道保护作用(Fig 5H-J)。
最终,该研究完整阐明了DPF2调控巨噬细胞炎症的分子机制:辐射或炎症刺激下,巨噬细胞中DPF2表达上调,通过表观遗传调控直接激活CACNA1D的转录,促进细胞膜表面L型钙通道的表达,增强胞外Ca²+内流,升高胞质钙浓度,进而激活下游p38/p44/42 MAPK信号通路,最终驱动巨噬细胞M1型促炎极化,分泌大量促炎因子,加剧肠道炎症损伤与黏膜屏障破坏;而DPF2缺失可阻断这一通路,抑制巨噬细胞的促炎活化,发挥肠道保护作用。
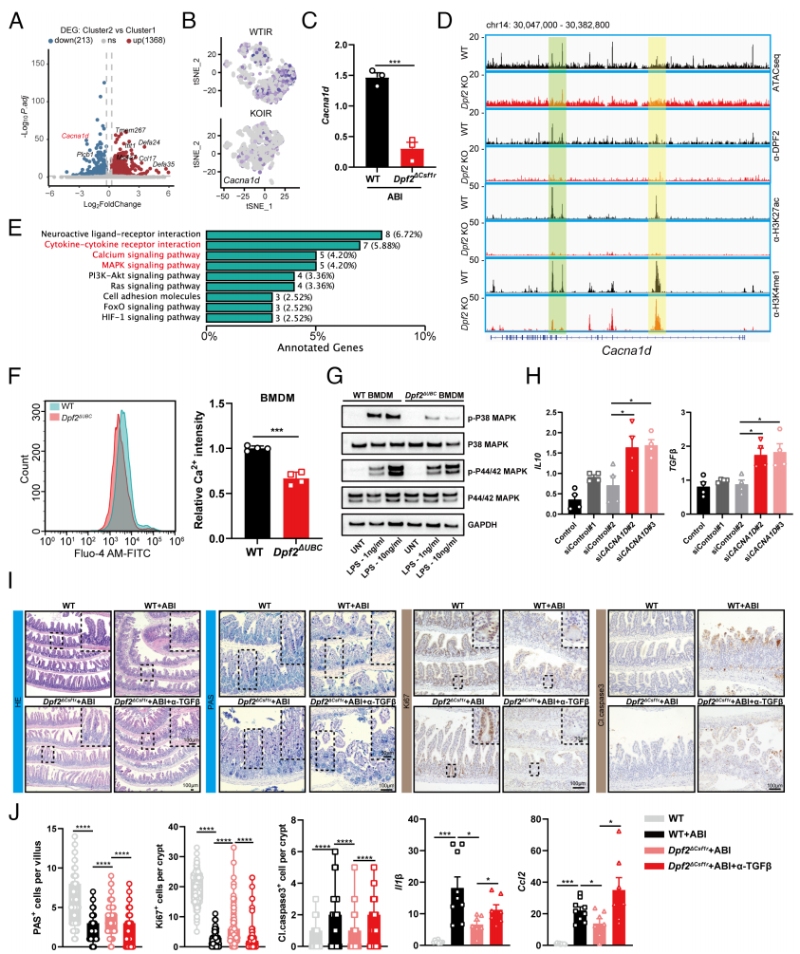
图5
DPF2/CACNA1D与临床肠道炎症正相关,靶向可促进肠类器官修复
为验证该机制在人类疾病中的相关性与转化价值,该研究完成了系统的人源样本验证与体外实验,为临床转化提供了坚实的证据:对GEO数据库中已发表的UC患者队列(GSE123141)进行了系统分析,该队列纳入了10例活动期UC患者与9例健康对照人群,对肠道CD14+/CD163+巨噬细胞进行了测序分析。DPF2 high/CACNA1D high巨噬细胞呈促炎M1表型,在KOIR空间数据中显著减少(Fig6A-C)。UC患者巨噬细胞DPF2、CACNA1D及促炎因子、钙转运通路显著升高(Fig6D)。该研究构建了人源结肠类器官,敲低DPF2或CACNA1D可显著恢复类器官形成能力(Fig6E-F)。临床放射性肠炎样本显示,高炎症组DPF2+、CACNA1D+、DPF2+CACNA1D+巨噬细胞比例显著更高(Fig6G-H)。
结果表明DPF2/CACNA1D与人类肠道炎症正相关,可作为辐射肠损伤与IBD潜在治疗靶点。
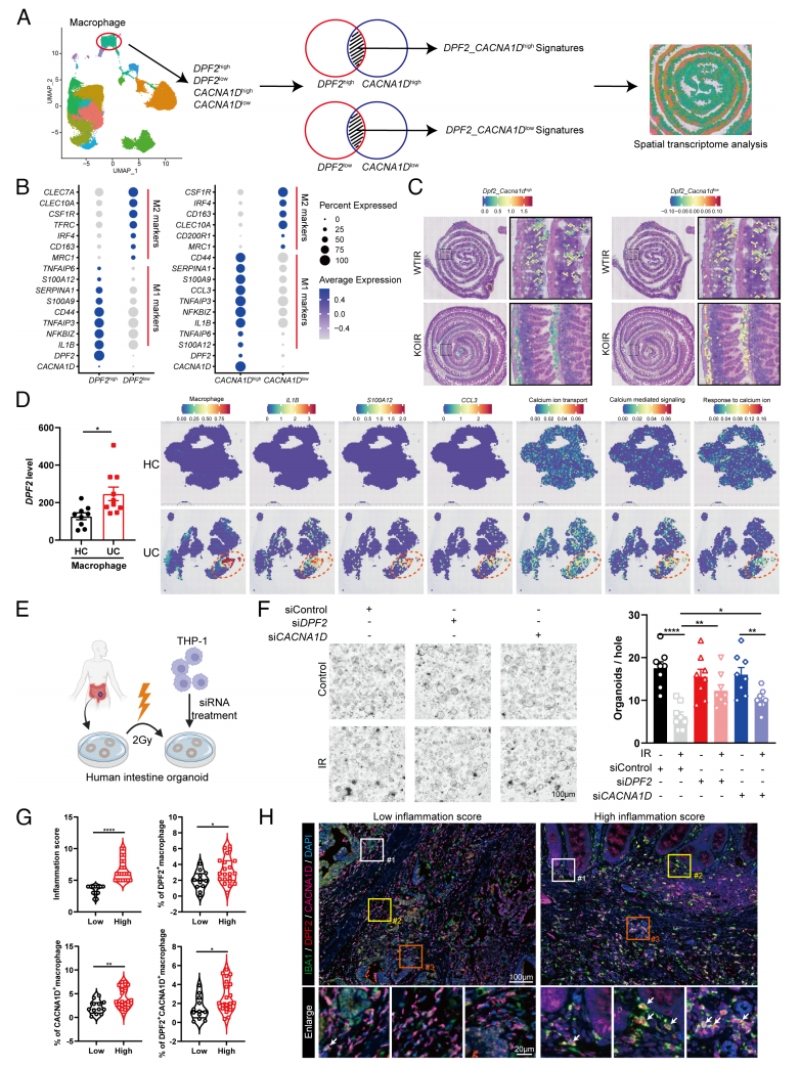
图6
研究总结
该研究突破性地揭示了SWI/SNF染色质重塑复合物亚基DPF2在肠道炎症损伤中的核心调控作用,首次完整阐明了DPF2-CACNA1D-钙信号-MAPK轴调控巨噬细胞炎症极化、驱动肠道辐射损伤与结肠炎进展的全新分子机制。
该研究不仅刷新了领域对染色质重塑复合物调控固有免疫炎症的基础认知,破解了巨噬细胞炎症极化的表观遗传调控谜题,更从免疫细胞-上皮细胞互作的角度,揭示了肠道炎症损伤的全新发病机制,为肠道辐射损伤、炎症性肠病提供了全新的早期诊断标志物与精准治疗靶点,具有里程碑式的科学价值与临床转化意义。
引用文献:
The SWI/SNF chromatin-remodeling subunit DPF2 regulates macrophage inflammation in intestinal injury via the CACNA1D-mediated MAPK pathway—《pnas》November 12, 2025.




 京公网安备 11011302003368号
京公网安备 11011302003368号