

2026年1月7日,Nature Plants在线发表了来自德国杜塞尔多夫海因里希·海涅大学的Rüdiger Simon团队的研究论文:“Imputation integrates single-cell and spatial gene expression data to resolve transcriptional networks in barley shoot meristem development”。

该研究通过创新的数据整合方法,将深度单细胞RNA测序数据与空间基因表达数据相结合,首次实现了在大麦组织中以细胞分辨率解析超过40,000个基因的表达图谱。该研究揭示了大麦从分生组织及器官起始细胞命运决定到特定花器官起始过程中的关键转录事件与时空轨迹,阐明了大麦花序和花器官发育的遗传调控基础,为未来精准定向改良大麦农艺性状提供了全新的研究工具和理论基础,具有重要的科学意义和应用价值。百迈客生物为该研究提供了转录组测序服务。
研究背景
禾本科植物的花序是复合结构,包含一系列作为干细胞巢的分生组织。这些分生组织在身份和寿命上各不相同,产生分支或分裂形成最终产生种子的花分生组织。在分生组织内部,特定的细胞类型由位置信息和基因调控网络的区域性活性所决定。然而,目前的技术难以获取基因表达谱的精确时空信息,从而限制了对这些局部微环境及复杂发育过程的理解。尽管已有一些单细胞转录组数据可用于禾本科物种,但要推断复杂组织内细胞的起源和命运,仍需依赖已知标记基因的表达谱进行间接推测。
研究结果
深度单细胞RNA测序数据
本研究首先通过深度单细胞RNA测序(scRNA-seq)技术,分析了参考品种“Golden Promise”在特定发育阶段的营养茎尖分生组织和幼穗细胞,获得了超过16,000个高质量单细胞的转录组信息,出于实验设计严谨的考虑,通过比较完整组织和原生质体组织的bulk RNA-seq数据去除了原生质体分离过程中诱导产生的差异基因,并鉴定出代表维管束、原套、内体层和分裂细胞等不同谱系的细胞集群。
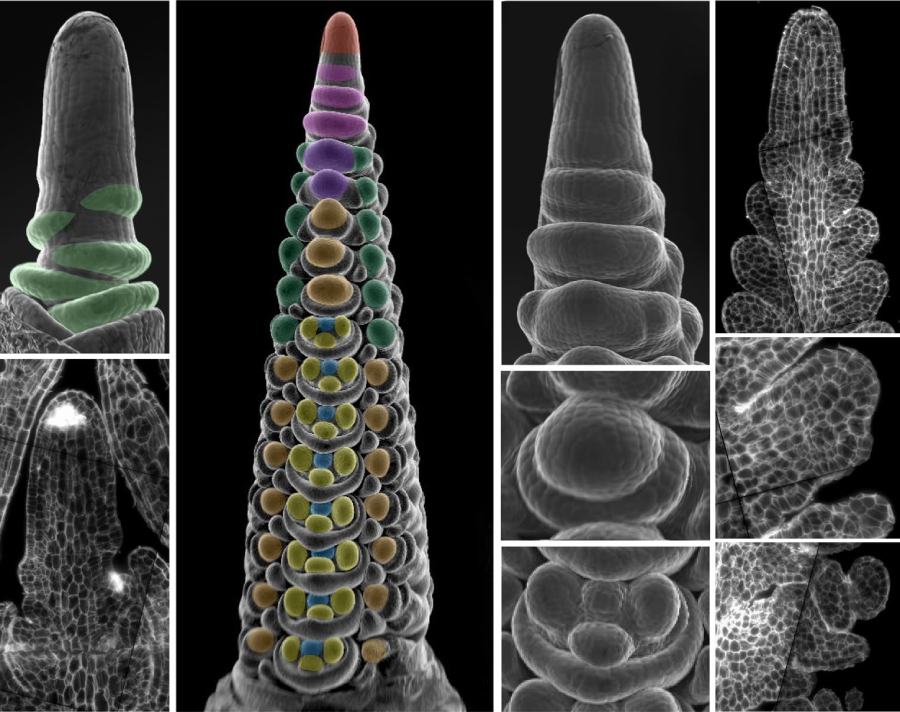
图1. 大麦发育尖端的器官形成
空间基因表达数据
然后,研究进一步通过高分辨率多重单分子RNA荧光原位杂交技术,在组织切片上以纳米级精度定位和定量了86个已知或预测标记基因的表达,并构建了空间表达矩阵。最终,研究者开发了一种定制化的数据插补方法,将空间有限的smRNA-FISH数据作为锚点,与全面的scRNA-seq数据集进行整合,成功地预测了组织切片上所有48,904个大麦基因在每个细胞中的表达水平,并构建了用户友好的在线图谱BARVISTA。这一系列工作说明,整合单细胞与空间转录组数据能够以前所未有的时空分辨率重建大麦分生组织发育的动态基因表达图谱。
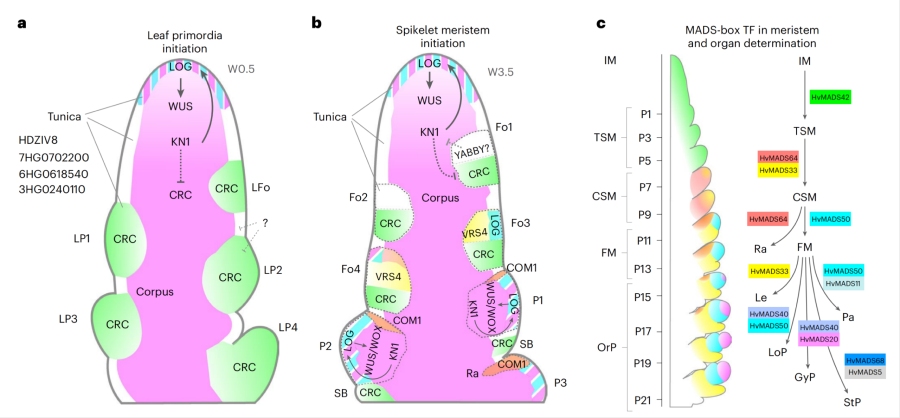
图2. 植物SAM中原基形成和分生组织结构的模型,以及发育中的花序示意图
研究总结
该研究的核心意义在于成功开发并验证了一种可靠的数据整合与插补策略,弥补了当前空间转录组技术通量有限和单细胞测序缺乏空间信息的鸿沟。所构建的BARVISTA在线数据库和虚拟显微切割工具,使研究人员能够直观地探索大麦发育过程中的基因表达模式,并精准解析特定细胞群体的表达特征。这项成果不仅为深入理解植物分生组织维持、器官起始和花发育的基因调控网络提供了强大框架,也为分子层面精准表征复杂突变体表型、鉴定关键调控因子开辟了新途径。
以上内容来源于iPlants,侵删




 京公网安备 11011302003368号
京公网安备 11011302003368号