

文章名称:Construction and Annotation of Ascosphaera apis Full-Length Transcriptome Utilizing Nanopore Third-Generation Long-Read Sequencing Technology
发表期刊:中国农业科学
发表时间:2020年11月
影响因子:2.302
研究背景
蜜蜂球囊菌(Ascosphaeraapis,简称球囊菌)是专性侵染蜜蜂幼虫的致死性真菌病原,引发的白垩病是长期危害养蜂生产的顽疾,不仅可导致蜜蜂幼虫的大量死亡,还能导致成年蜜蜂数量的锐减以及蜂群群势和蜂产品产量的骤降。目前,球囊菌的基因组注释信息尚不完善,高质量参考转录组匮乏,严重限制了球囊菌的组学和分子生物学研究。
材料和方法
球囊菌菌株由福建农林大学动物科学学院(蜂学学院)蜜蜂保护实验室分离、纯化和保存。纯化得到的纯净菌丝样品和孢子样品经液氮速冻后迅速转移到-80℃超低温冰箱保存备用。利用纳米孔长读段测序技术对球囊菌的纯化菌丝(Aam)和纯化孢子(Aas)分别进行测序,将高质量的三代测序数据混合后用于构建全长转录组,并通过比对主流数据库进行功能注释,同时对球囊菌的长链非编码RNA(longnon-codingRNA,lncRNA)进行鉴定和分析。
结果
1、纳米孔测序数据质控
球囊菌菌丝和孢子的纳米孔测序分别得到6321704和6259727条原始读段,N50分别达到1094和1157bp,平均长度分别为992和1047bp,长的长度分别为9421和13060bp(表1)。来源于Aam和Aas的原始读段的长度分布介于1-10kb以上,其中分布reads数多的长度均为1kb(图1-A、1-B);原始读段的Q值分布介于Q6-Q15,分布reads数多的质量值分别为Q9和Q11(图1-C、1-D)。
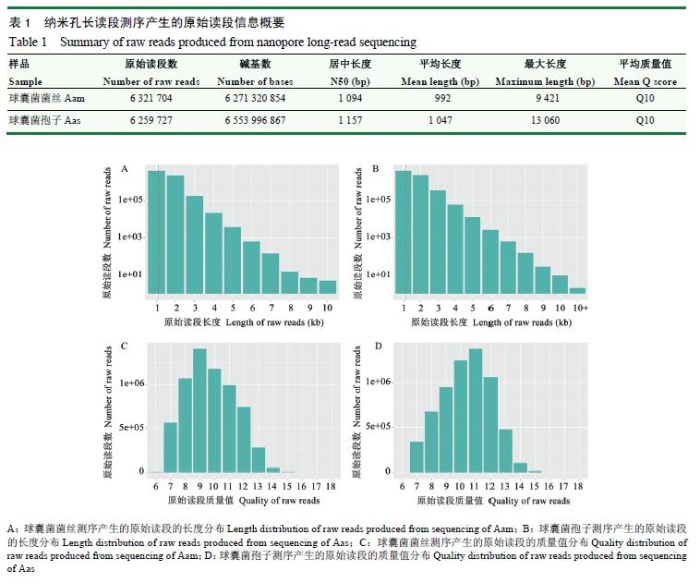
图1球囊菌菌丝和孢子纳米孔长读段测序的原始读段长度和质量值分布Fig.1Lengthandqualitydistributionofrawreadsgeneratedfromnanoporelong-readsequencingofA.apismyceliumandspore
2、全长转录本的鉴定和分析
进一步过滤冗余全长有效读段,分别得到9859和16795条非冗余全长转录本,N50分别达到1482和1658bp,平均长度分别达到1187和1303bp,长的长度分别为6472和6815bp(表2);上述非冗余全长转录本的长度介于1-7kb,其中分布在1kb的全长转录本数多。进一步对Aam和Aas的非冗余全长转录本进行Venn分析,结果显示有6512个非冗余全长转录本为菌丝和孢子所共有,分别有3347和10283个非冗余全长转录本为二者特有(图2-A)。
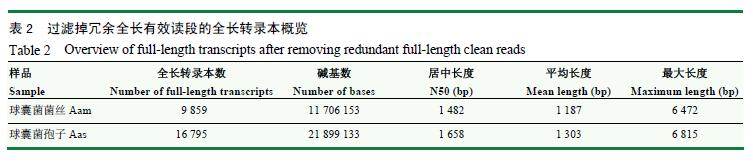
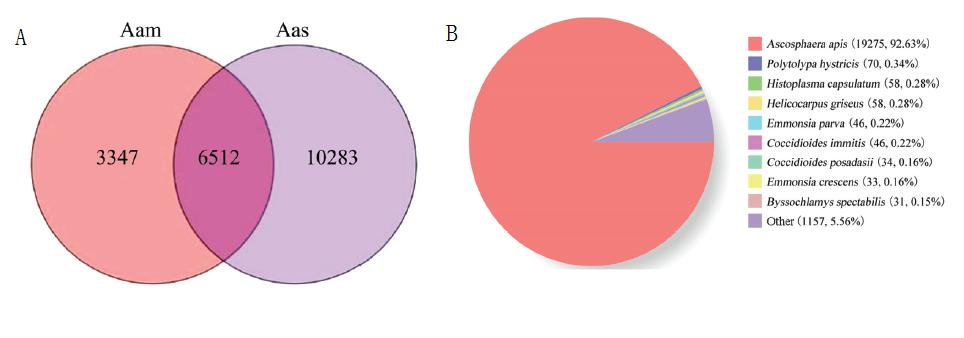
图2球囊菌菌丝和孢子全长转录本的Venn分析(A)、全长转录本的Nr数据库注释(B)Fig.2Vennanalysisoffull-lengthtranscriptsinA.apismyceliumandspore(A)、Nrdatabaseannotationoffull-lengthtranscripts(B)
3、全长转录本的数据库注释
在球囊菌菌丝和孢子中共鉴定出20142条全长转录本,数据库注释结果显示,分别有20809、11151、17723、12164、11340和9833全长转录本可注释到Nr、KOG、eggNOG、Pfam、GO和KEGG数据库。注释全长转录本数量多的物种是球囊菌、Polytolypahystricis和荚膜组织胞浆菌(Histoplasmacapsulatum)(图2-B)
4、lncRNA的鉴定及分析
利用CPC、CPAT、CNCI和Pfam4种方法依次鉴定出1906、1682、750和648条lncRNA,四者的交集为648个(图3-A);其中基因间区lncRNA(longintergenicRNA,lincRNA)、反义链lncRNA(anti-senselncRNA)和正义链lncRNA(senselncRNA)的数量分别为480、119和49个(图3-B)。
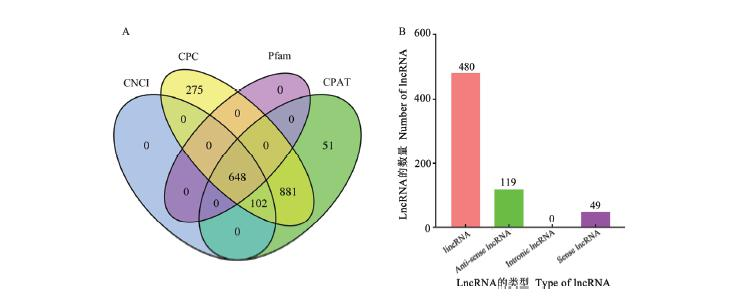
图 3 球囊菌 lncRNA 的数量(A)和种类(B) Fig. 3 Number (A) and type (B) of A. apis lncRNAs
总结
构建和注释了球囊菌的高质量全长转录组,为探究球囊菌转录组的复杂性、完善参考基因组的序列和功能注释信息以及深入开展球囊菌可变剪接体的功能研究提供了关键依据。
深度挖掘数据和拓展
同期作者利用纳米孔全长转录组测序数据对蜜蜂球囊菌(Ascosphaeraapis)和另一蜜蜂真菌病原东方蜜蜂微孢子虫(Nosemaceranae)的现有参考基因组在结构功能注释上进行了较好的完善,同时也对基因的可变剪接(alternativesplicing,AS)和可变多聚腺苷酸化(alternativepolyadenylation,APA)进行解析。通过gffcompare软件将全长转录本与参考基因组注释的转录本进行比较,对基因组注释基因的非编码区向上游或下游延伸,修正基因的边界。利用MISA软件鉴定长度在500bp以上的全长转录本的简单重复序列(simplesequencerepeat,SSR)位点信息。使用Blast工具将鉴定到的新基因和新转录本比对Nr、KOG、eggNOG、GO和KEGG数据库,从而获得功能注释。通过Astalavista软件鉴定基因的AS事件类型,统计分析可变剪切的结果。采用TAPISpipeline对基因的APA位点进行鉴定,得到APA的位点信息。分别利用CPC、CNCI、CPAT、Pfam4种方法对长链非编码RNA(longnon-codingRNA,lncRNA)进行预测,取四者的交集作为高可信度的lncRNA。研究结果较好地优化了现有的东方蜜蜂微孢子虫和蜜蜂球囊菌参考基因组已注释基因的结构和功能注释信息,并补充和注释了大量参考基因组未注释的新基因和新转录本,同时也为其他真菌的AS和APA研究提供了有益的思路和方法借鉴。




 京公网安备 11011302003368号
京公网安备 11011302003368号