

在生物学的研究中,有一个常用的方法,就是通过比较分析获取有用的信息和知识。我们现在最常见的比对是蛋白质序列之间或核酸序列之间的两两比对,通过比较两个序列之间的相似区域和保守性位点,寻找二者可能的分子进化关系。我们在进行功能注释的时候也是基于序列相似 功能相似这样的一个原理来进行的。
目前BLAST (Basic Local Alignment Search Tool)是现在应用最广泛的序列相似性搜索工具,它运用某种特定的数学模型或算法,找出两个或者是多个序列之间的最大匹配碱基或氨基酸残基数,比对的结果反映了算法在多大程度上提供序列之间的相似性关系及它们的生物学特征。BLAST因其快速及比对精度高而被广泛应用于双序列或多序列比对。
大家非常熟悉的是NCBI上面的BLAST比对,还记得小编读研期间在师兄的谆谆教诲下学到的第一个技能就是BLAST比对,可见它的重要性及广谱性!因为大家对NCBI上的BLAST非常熟悉,这里我就不做过多的介绍了。今天跟大家分享的是一款可视化的比对工具:百迈客云(BMKCloud)—BLAST。
序列比对与NCBI采用的都是第三方BLAST,其数学模型或算法都是一样的。可能大家要问了,既然都一样,那我为什么要选择BMKCloud呢?在这里我要隆重介绍介绍,BMKCloud优势如下:
2、无限制:我们可以比对很多的序列,而NCBI上是有限制的,超过上限将不能运行比对任务。
很多时候我们拿到转录组的测序数据,需要在海量的数据里找到自己关注的基因。我们可以通过关键词进行功能注释的搜索,还有常见的方法是我们关注的 基因,可能在别的物种或者是近缘物种已有报导。针对这种情况我们就可以通过已知的基因序列运用BLAST比对在我们的测序数据里找同源序列。
那这个时候你需要的是什么?
大声告诉我~~
对,就是百迈客云(BMKCloud)小工具—BLAST!

依然是非常简单的操作步骤:
1.登录百迈客云首页(www.biocloud.net)——分析——工具
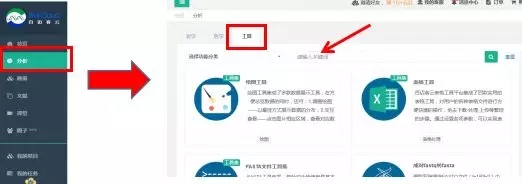
2.在对话框中输入“BLAST”,Enter键进入工具使用界面
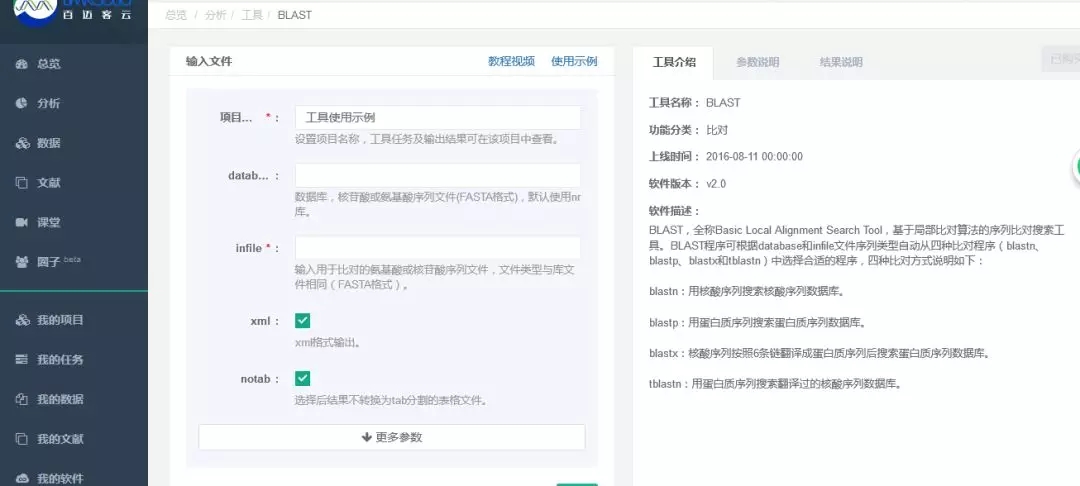
3. 这里有几个输入框或勾选框,主要包括:
1、 项目名称
2、database :用于建库的核苷酸或氨基酸序列文件,默认使用NR库(FASTA格式)。
3、 infile :用于比对的序列文件,可以为核酸序列或者氨基酸序列。
4、 expect :非负数,默认的e值为10,E值越小,说明比对结果越可信。一般做基因的注释,与已知数据进行同源性搜索,通常设定为1e-5或 更小的值。
5、 qcov :0-100,默认为80,查询序列与目标序列的覆盖率。
其中两个必填输入框
项目名称:即根据自己需求对本次BLAST进行命名描述,以便于后期查看结果与
infile :在输入比对的序列文件时,有三种方式,包括选择云文件、浏览本地文件或拖拽文件
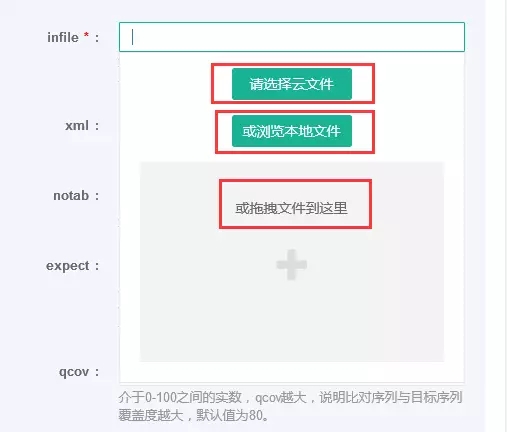
另外,之前提到BMKCloud很重要的一个优势在于可以拿任何fasta文件当做比对的数据库,因此在“database”这一输入框中大家就可以选择自己的转录组数据作为比对的参考数据库。
4.完成项目名称和fasta文件输入之后,点击提交即可运行比对任务。
BMKCloud还有很多既操作简单又实用的小工具,如果你正对着一组数据苦恼的话,那就来BMKCloud了解一下吧,可能会有很多惊喜发现啊!




 京公网安备 11011302003368号
京公网安备 11011302003368号