


酵母以其易于基因改良且生长迅速的特点,成为学术研究和工业生产的利器。近些年,随着测序技术的发展,更是晋升为三代测序技术的团宠。2017年发表在Genome Research(IF=10.101)上的“The dynamic landscape of fission yeast meiosis alternative-splice isoforms”(请点击阅读原文下载)在学术圈广为流传,是利用 PacBio 研究可变剪接的经典案例。
今年酵母搭乘 Nanopore 再发2篇高分文章(请点击下载原文),从基因组、转录组体验长片段测序的利好。
Nanopore 测序到底怎么样?可以做定量吗?和 PacBio 相比如何?
这篇“Complete genomic and transcriptional landscape analysis using third-generation sequencing: a case study of Saccharomyces cerevisiae CEN.PK113-7D”告诉您!(请点击下载原文)
时间:2018 年 4 月
杂志:Nucleic Acids Research
影响因子:11.561
材料和方法
样品:酵母菌种 CEN.PK113-7D
测序方案:
◈ 全基因组测序:
☻ PacBio,Sequel,~4900Mb,N50为8700bp,约408X(PRJNA398797,SRP116559)
☻ Nanopore,MinIon,~830Mb,N50 为 12500bp,约 69X(PRJNA398797, SRP116559)
☻ Illumina,HiSeq 2000,先前研究序列(SRS307298)
◈ 全长转录组测序:
☻ Nanopore,MinIon,direct RNA,葡萄糖生长条件和乙醇生长条件,各 4 个 生物 学重复(PRJNA398797, SRP116559 )
☻ Illumina,HiSeq 2000,先前研究序列(SRS307298)
主要结果
1、基因组数据特征分析
相比于 PacBio,MinIon 能产出更长的序列,但是 PacBio 测到的序列数量是 MinIon 的 5.6 倍左右。大部分的序列在两个平台都能检测到,个别特有的序列可能和实验阶段的片段筛选有关。
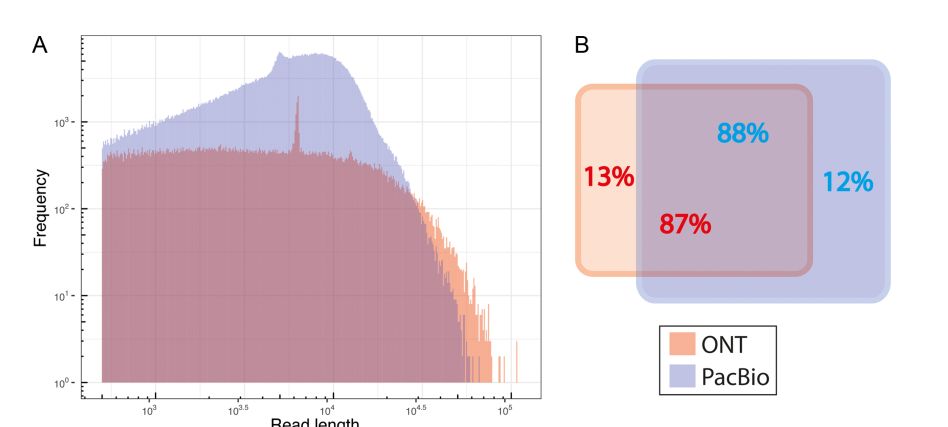
图 1 MinIon and PacBio 数据特征统计
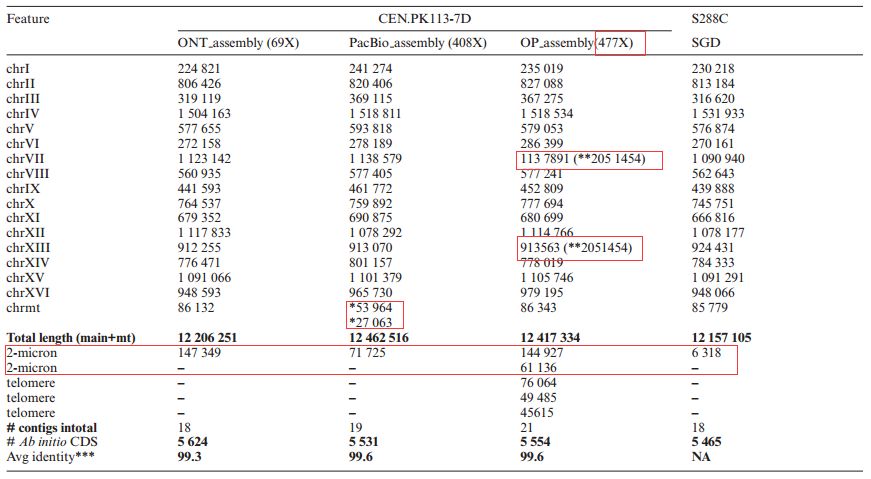
对于不同测序平台得到的基因组数据,采用 3 种组装方式:只用 MinIon 得到的数据进行 de nove组装得到 ONT_assembly;只用 PacBio 得到的数据进行 组装得到 PacBio_assembly;用 MinIon和 PacBio 得到的数据共同进行 de nove 组装得到 OP_assembly。然后使用 Illumina 的数据对组装结 果进行校正。三种组装方式得到的 2μm 质粒都比已知的 S288C 酵母的 2μm 质粒更长,而ONT_assembly 的组装结果很好,刚好组装出 16 条染色体,1个线粒体序列,1个质粒序列;PacBio_assembly 组装的线粒体序列是断裂开的两段;OP_assembly 组装出的 contigs 最多,多出 3个端粒 DNA 片段,2 个质粒片段,而且组装时把 VII 染色体和 XIII 染色体的端粒区域连接在了一 起。 总体来看,三种组装方式得到的基因组很相近,由于 OP_assembly 的测序深度最高,与 S288C酵母的平均比对率达到了 99.6%。
表 1 三种组装方式的数据比较
2、全长转录组数据特征分析
二次生长(diauxic growth)是指微生物可以利用一种营养物质产生代谢产物,当原始营养 物质被利用完后,可以进一步利用其代谢产物生长繁殖,产生新的代谢产物。最典型的二次生长 现象就是酵母菌的二次生长,即在糖源丰富时,酵母将葡萄糖糖转化为乙醇,当葡萄糖耗尽后, 酵母可以进一步将乙醇氧化为乙酸,维持生长。
分别对葡萄糖条件和乙醇条件下生长的酵母进行全长转录组测序,葡萄糖生长条件下的酵母 共计获得~509 MB(59X)数据量,包含约 530,000 高质量 reads,其中 N50 为 1150bp;乙醇条 件下的酵母共计获得~623 MB(72X)数据,约 623000 高质量 reads,其中 N50 为 1263 bp。该 技术的比对率为 88%,错误率为 12%,其中超过 70%的转录本为全长转录本,鉴定长度最长转录 本超过 5kb。全长转录本的比例随着转录本长度增加而减小,与转录本的表达量没有明显关系。有22 个转录本是明显高表达的,比如 丰度最高的转录本之一, 的同源 基因,在两个培养条件下表达量都很高;在乙醇条件下特异高表达一些与热休克蛋白、氧化胁迫 相关的基因,其中 编码柠檬酸合酶,说明此时激活了乙醛酸途径;在葡萄糖培养条件下,与 有氧呼吸、核糖体相关的基因表达量较高,符合此条件下酵母生长更快的特征。
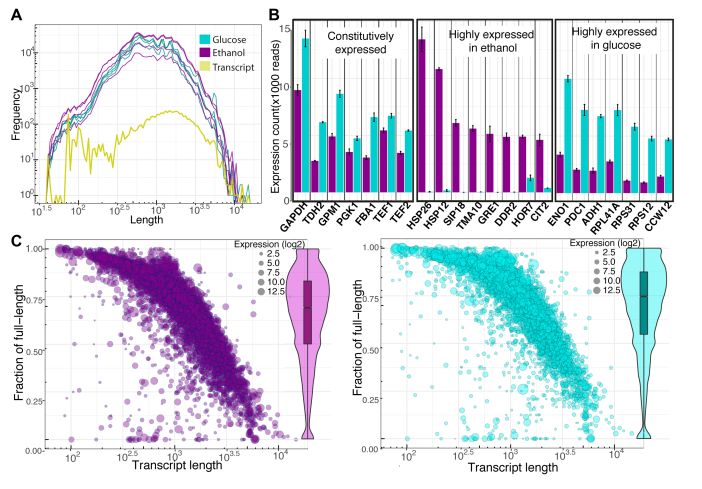
图 2 转录组数据特征统计
3、全长转录组数据差异筛选
通过主成份分析(PCA)发现两组数据有明显的差异,PC1 的贡献率达到 90%。使用 DESeq2做差异分析并对差异基因做 GO 富集分析,结果显示葡萄糖培养条件下的上调基因富集到了与转 录、翻译过程相关的 GO term,与葡萄糖培养条件下生长更快的表型吻合。在乙醇培养条件下, 上调基因主要富集到了 TCA 循环,乙醛酸通路,线粒体电子传递方面。同时,由于营养物质消耗、 毒性代谢物积累,在乙醇条件下很多与胁迫响应,分解代谢,β氧化相关的基因表达量上调。
4、转录组:MinION VS Illumina
由于 Illumina 产出的是短片段,比对上基因组的 reads 数比 MinION 要多 10 倍左右。比对上的碱基总数可以反映测序深度,统计发现 MinION 约有 0.5G 数据,测序深度 64X,Illumina 约有1G 数据,测序深度 118X。(图 3E)而在平均覆盖率的统计中发现,两种平台非常相似。(图 3F) 可见,在相似的覆盖率需求下,MinION 需要的数据量更少。
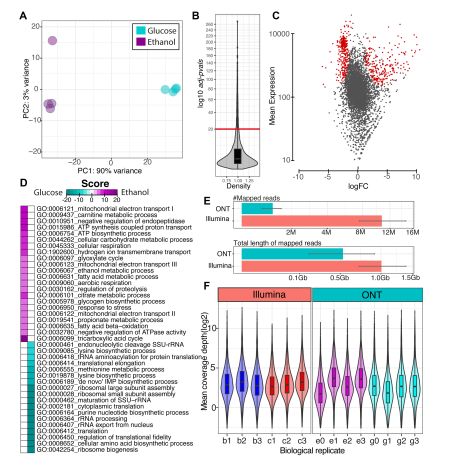
图 3 转录组差异分析
5、转录组数据结构分析
MinION 的数据中,我们发现相邻的 2 个基因 PTH1 (CENPK0H0281W) 和 ERG9 (CENPK0H0282W) 转录在一个转录本中,有大量的 reads 跨越了基因间区。而 Illumina 的比对结果中, 在两个 ORFs 之间的区域,比对到的 reads 不是完全覆盖的,这种低置信度的信息很可能被忽略。
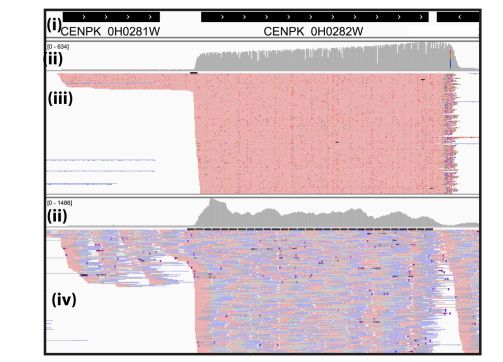
图 4 THI1 和 ERG9 比对结果可视化
总结
本文将第三代测序技术与成熟的生物信息学分析流程结合起来,成功组装出真核生物的基因组。长片段测序使高质量、完整的真核基因组序列的重新组装成为可能,这为比较基因组学奠定了坚实的基础。Nanopore 测序能够准确测定编码的 mRNA 位置,基因表达量,以及转录本的结构。我们相信,Nanopore 测序将成为未来基因组和转录组重要方式。应该注意的是,本文研究的是基因组简单的酵母,在处理高等生物(动物和植物)的基因组和转录组时,需要更高的测序深度和更长的读取。
今年8月份牛津纳米孔公司与百迈客公司达成长期合作,已引入Oxford Nanopore平台,拥 有 MinION、GridION X5 和 PromethION 三种型号全套纳米孔测序仪。
在这盛世华诞来临之际
我们给科研大咖奉上钜惠大礼!!!

想尝试新技术!
想快速发文章!
想发高分文章!
请联系我们免费获取文章思路设计方案!





 京公网安备 11011302003368号
京公网安备 11011302003368号