
LinkageMapView是一个用R编写的免费的包,可以绘制遗传连锁图谱和QTL图谱,我们在拿到遗传图谱之后,可以通过此功能来对图谱进行可视化,展示的形式也是多种多样的,还可以在图片上增加QTL的内容,最方便的是可以直接将R/qtl的输出作为LinkageMapView的输入,也可以接收txt和csv格式。
1.下面具体介绍一下包的安装和使用
安装的命令:
LinkageMapView包中主要有一个函数——lmv.linkage.plot,包括的参数非常多,在说明文档中仅仅是参数说明就用了4页,所以我们了解一些常用的即可。 2.下面进行实例介绍 1、最简单的图谱绘制Install.packages(“LinkageMapView”)
library(LinkageMapView)
代码如下:
library(qtl)
data(hyper)
lmv.linkage.plot(mapthis=hyper,outfile=”a.pdf”,mapthese=c(1,4,6,15),main=”genetic map”)
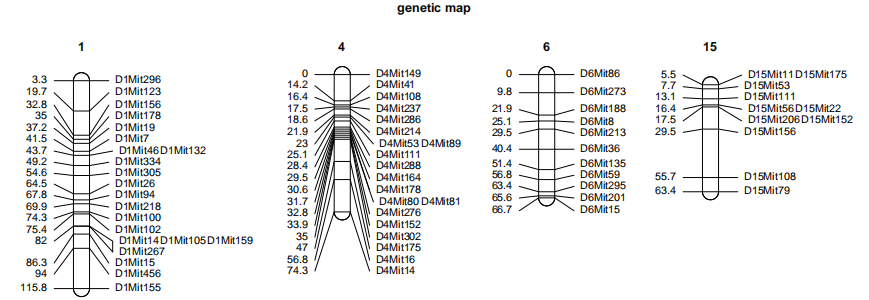
使用qtl包中hyper数据,mapthis是lmv.linkage.plot函数输入的数据集参数,第一列是连锁群编号,第二列是图距信息,第三列是标记名称,此参数为必给的参数,outfile是输出的pdf图片名称,必给的参数,mapthese用于选择绘制那些连锁群,可选参数,不设置的话,会默认将所有连锁群都绘制,main设置标题。
2、变更部分标记名称颜色
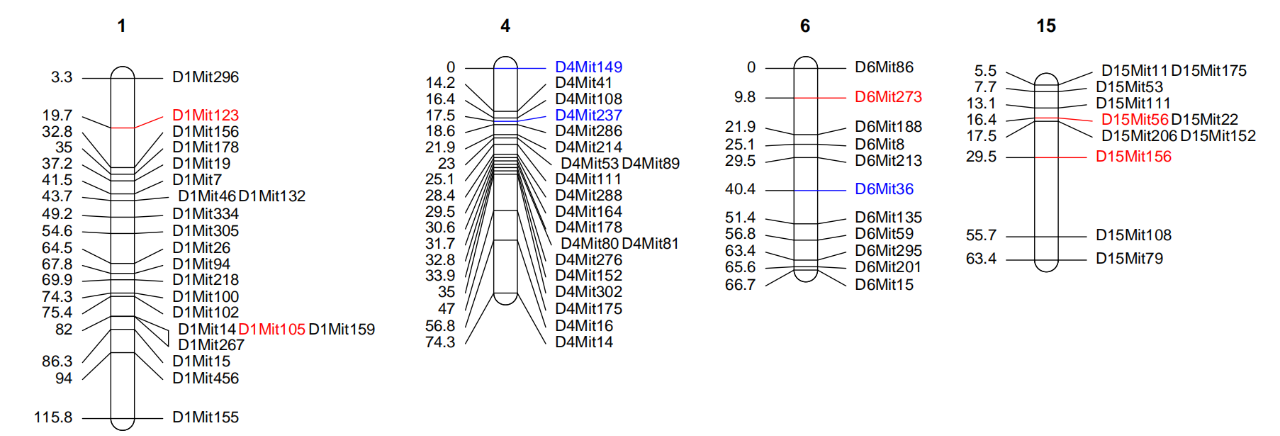
代码如下:
flist[[1]] <- list(locus = c(“D1Mit123″,”D1Mit105″,”D6Mit273″,”D15Mit56″,”D15Mit156”),col=c(“red”))
flist[[2]] <- list(locus = c(“D4Mit149″,”D4Mit237″,”D6Mit36”),col=c(“blue”))
lmv.linkage.plot(hyper,”b.pdf”,mapthese=c(1,4,6,15),markerformatlist=flist)
先创建一个有两个成分的列表,只想展示一个颜色就用一个成分的列表。locus给标记名称的向量,col给颜色的向量。Markerformatlist给创建的列表。
3、颜色设置
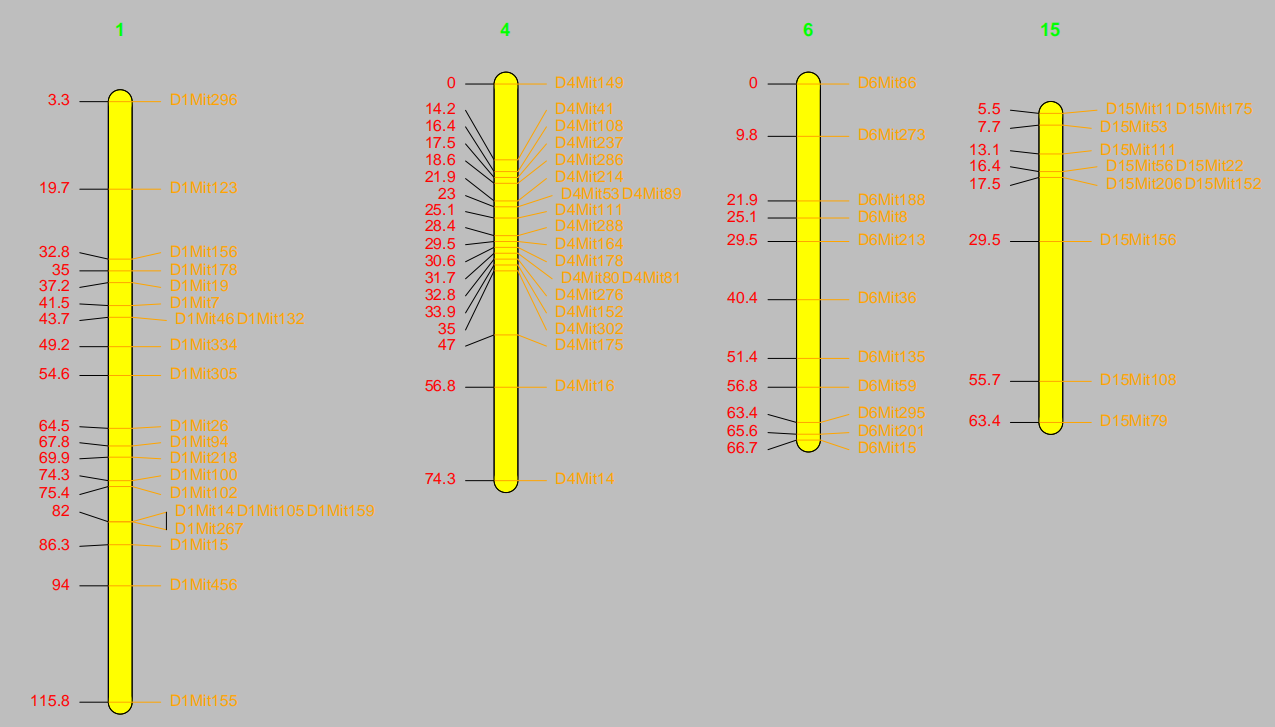
代码如下:
lmv.linkage.plot(hyper,”c.pdf”,
mapthese=c(1,4,6,15),
pdf.bg=”grey”,col.lgtitle=”green”,
lg.col=”yellow”,lcol=”red”,rcol=”orange”)
pdf.bg:pdf背景色
col.lgtitle:连锁群名称颜色
lg.col:连锁群柱子颜色
lcol:遗传位置label的颜色
Rcol:标记名称label的颜色
4、字体、字号设置
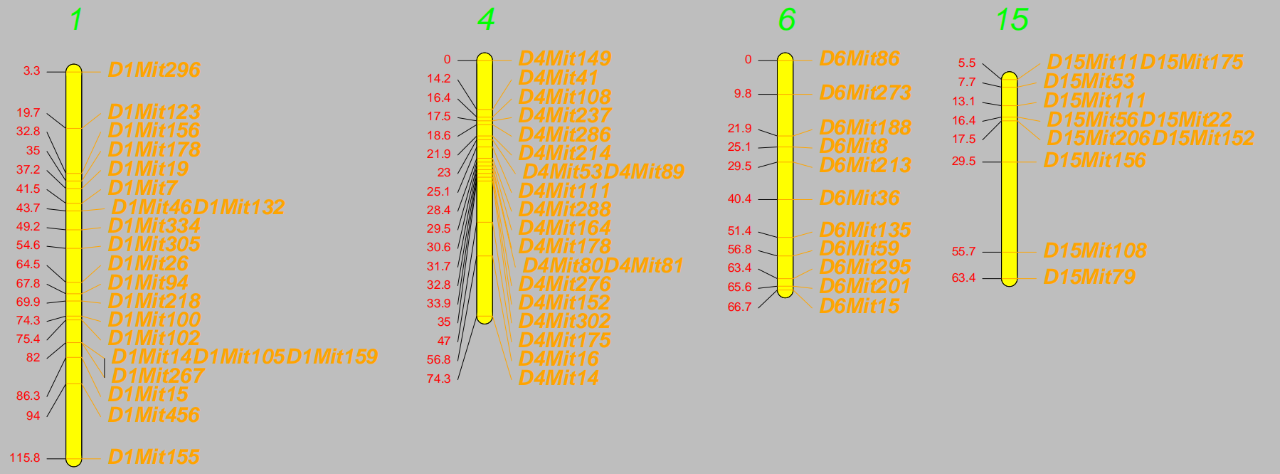
代码如下:
lmv.linkage.plot(hyper,”d.pdf”,mapthese=c(1,4,6,15),pdf.bg=”grey”,col.lgtitle=”green”, pdf.height=8,lg.col=”yellow”,lcol=”red”,rcol=”orange”, lfont=1,lcex=1.2,rfont=4,rcex=2,font.lgtitle=3,cex.lgtitle=3)
lfont:遗传位置label的字体设置,1代表纯文本,2代表粗体,3代表斜体,4代表粗斜体
lcex:遗传位置label的字号大小设置
rfont:标签label的字体设置
rcex:标签label的字号大小设置
font.lgtitle:连锁群编号字体设置
cex.lgtitle:连锁群编号字号大小设置
5、部分染色体柱子上色
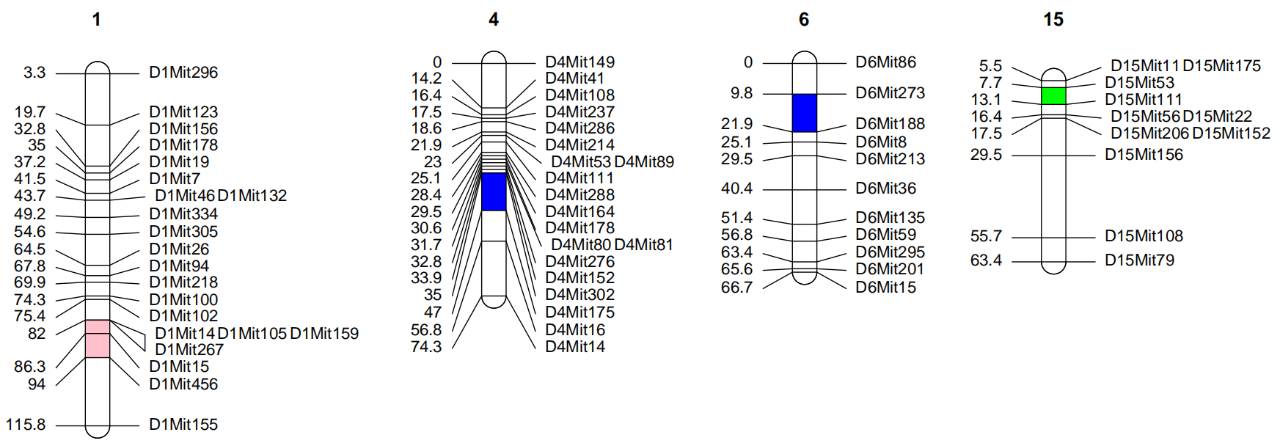
代码如下:
chr = c(1, 4, 6, 15)
s = c(82,35,9.8,7.7)
e = c(94,47,21.9,13.1)
col = c(“pink”,”blue”,”blue”,”green”)
sectcoldf <- data.frame(chr, s, e, col,stringsAsFactors = FALSE)
lmv.linkage.plot(hyper,”e.pdf”,mapthese=c(1,4,6,15), sectcoldf=sectcoldf)
sectcoldf为连锁群柱子部分上色参数,上色信息以data.frame格式储存,每一行是一个上色区域,第一列是连锁群编号,第二列是对应连锁群上色起始,第三列是上色终止,第四列是对应的颜色。
6、标签显示形式设置
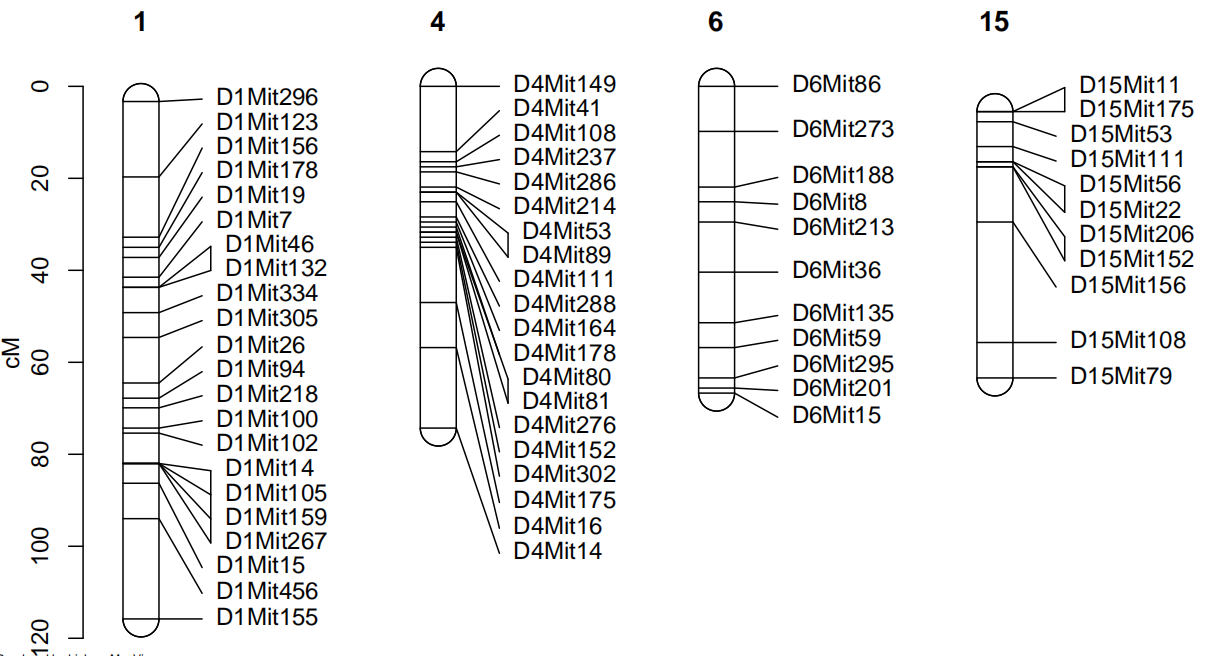
代码如下:lmv.linkage.plot(hyper,”f.pdf”,mapthese=c(1,4,6,15),ruler=TRUE,maxnbrcolsfordups = 1)
Maxnbrcolsfordups 表示标记label用几列表示,默认3.
7、相同标记名称连线
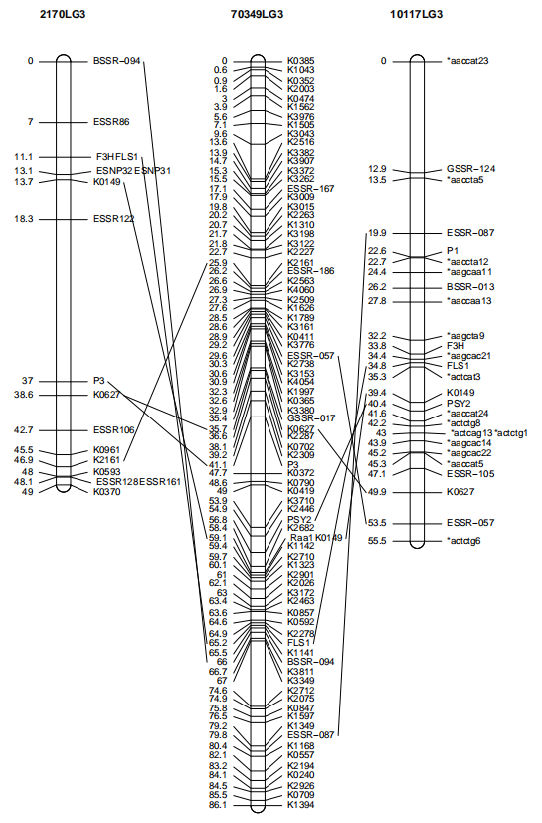
代码如下:
data(carrot)
lmv.linkage.plot(mapthis = carrot,outfile = “g.pdf”)
函数会自动将连锁群之间标记名一样的标记进行连线,autoconnadj=FALSE可以关闭此功能
8、标记名称不同连线
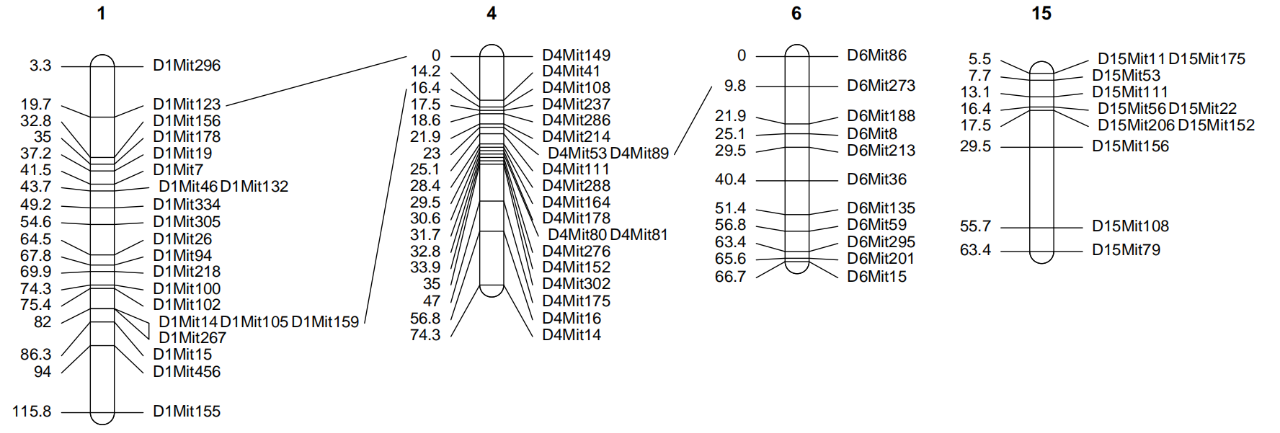
代码如下:
fromchr = c(1,1,4)
fromlocus = c("D1Mit123","D1Mit105","D4Mit53")
tochr = c(4,4,6)
tolocus = c("D4Mit149","D4Mit108","D6Mit273")
conndf <- data.frame(fromchr,fromlocus,tochr,tolocus,stringsAsFactors = FALSE)
lmv.linkage.plot(hyper,"h.pdf",mapthese = c(1,4,6,15),conndf = conndf)需要连线的标记对以data.frame存储,每行一对连线,第一列是线一端的起始连锁群编号,第二列是起始的标记名称,第三列是线另一端终止连锁群编号,第四列是终止的标记名称。9、翻转连锁群顺序
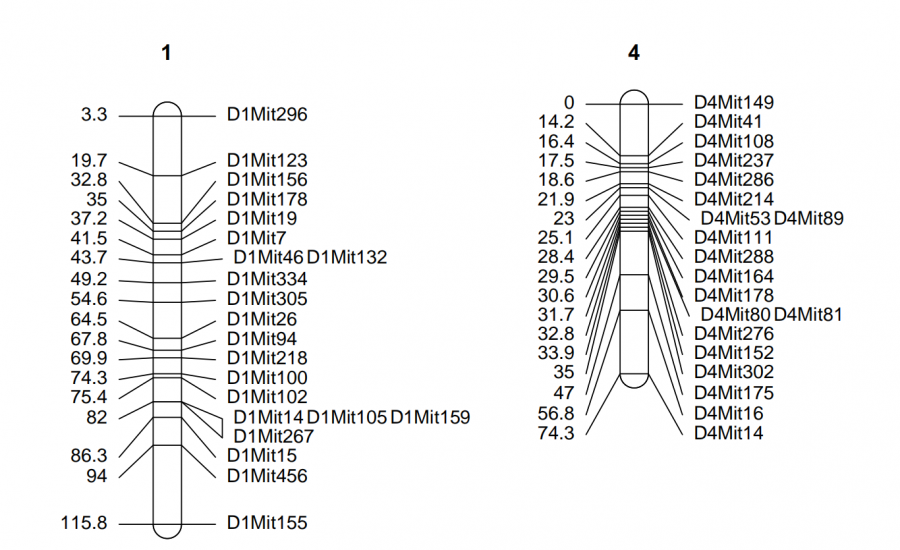
图一为原始图
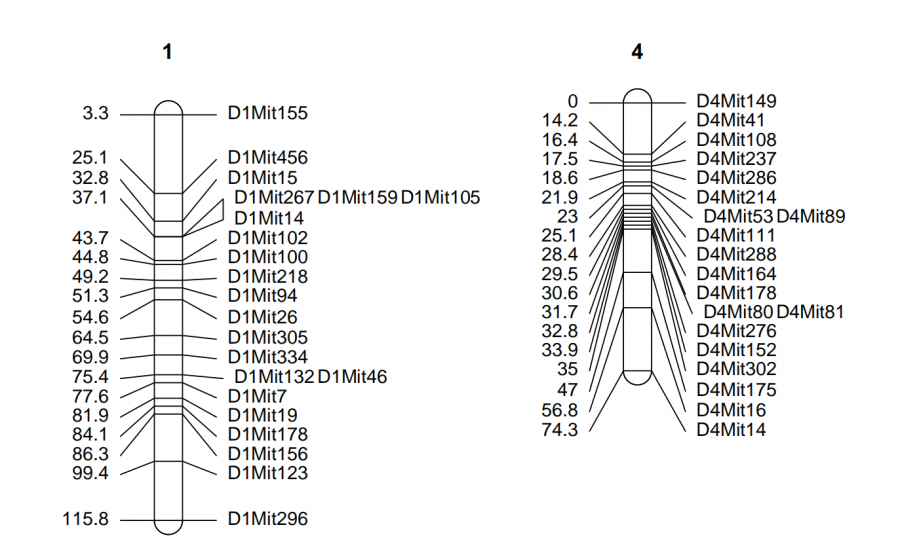
图二为翻转图
代码如下:
lmv.linkage.plot(hyper,”i.pdf”,mapthese = c(1,4),revthese = “1”)
revthese参数后面接需要翻转的连锁群编号
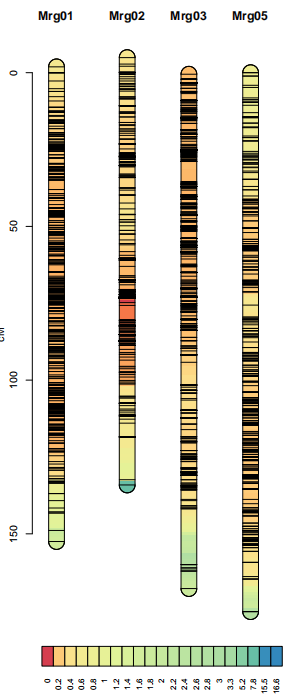
10、密度图谱绘制
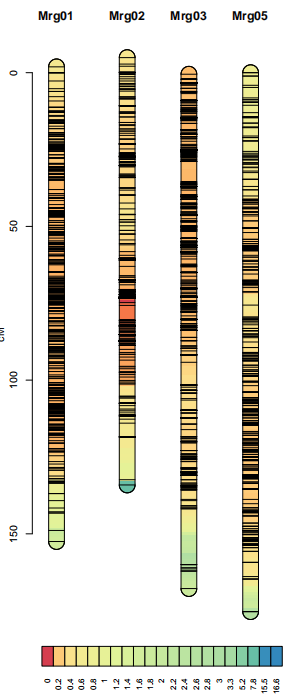
代码如下:
data(oat)
lmv.linkage.plot(oat,”j.pdf”,mapthese=c(“Mrg01″,”Mrg02″,”Mrg03″,”Mrg05”),denmap=TRUE)
denmap是控制是否画密度图的开关,画密度图的同时不显示标记名称和遗传位置信息。
11、QTL绘制
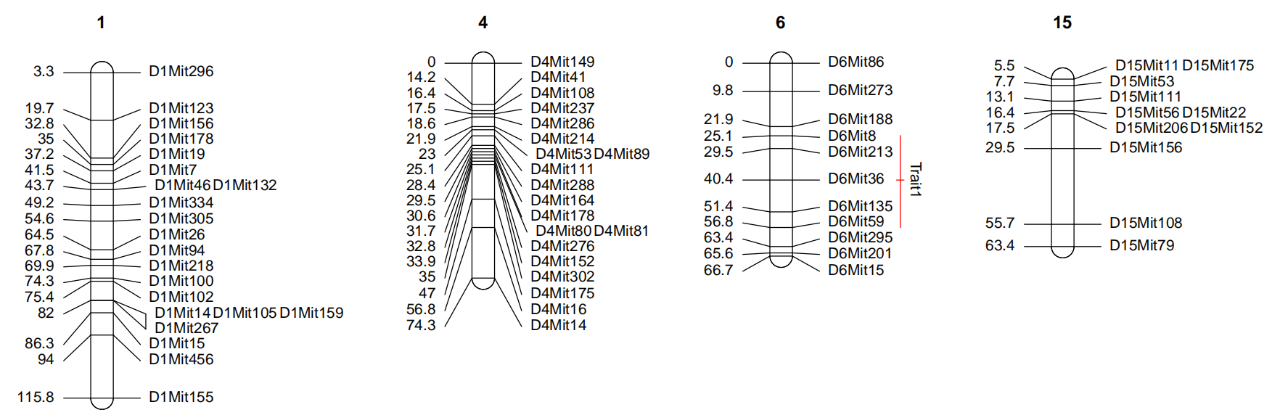
qtldf <- data.frame(
chr = character(),
qtl = character(),
so = numeric(),
si = numeric(),
ei = numeric(),
eo = numeric(),
col = character(),
stringsAsFactors = FALSE
)
qtldf <- rbind(qtldf,
data.frame(
chr = “6”,
qtl = “Trait1″,
so = 25,
si = 40.4,
ei = 40.4,
eo = 56.8,
col=”red”
))
lmv.linkage.plot(hyper,”k.pdf”, mapthese=c(1,4,6,15),qtldf = qtldf)
qtldf参数后接qtl信息,qtl信息存储在data.frame格式的qtldf里,一行一个qtl信息,第一列是连锁群编号,第二列是qtl名,第三列是qtl外区间起始,第四列是qtl内区间起始,第五列是qtl内区间终止,第六列是qtl外区间终止,第七列是颜色。
以上是LinkageMapView包常见的几个用法,LinkageMapView包可以接收R/qtl的输出结果也可以单独整理成本文输入,输入灵活,并且可以绘制高密度的图谱,更适用于现在的高通量测序下构建的遗传图谱。感兴趣的同学,赶快去试试吧!如果您还有什么疑问,也欢迎点击下方按钮联系我们,我们将免费为您设计文章思路方案。




 京公网安备 11011302003368号
京公网安备 11011302003368号