
想搭载nanopore三代测序新技术科研快车的你,get到研究套路了吗?ONT重测序和ONT全长转录组测序,双剑合璧!
Long read sequencing reveals a novel class of structural aberrations in cancers:identification and characterization of cancerous local amplifications.bioRxiv.2019
研究背景
国际癌症基因组协作组(International Cancer Genome Consortium,ICGC)和癌症基因组图谱(The Cancer Genome Atlas,TCGA)等已经揭示了多种癌症的致病性突变,其中包括研究较深入的肺腺癌LUAD。超过一半的LUAD病例具有EGFR和KRAS基因特征性点突变,或者ALK、RET和ROS1的基因融合,并被应用于靶向治疗策略指导。但,大约20-30%的肺腺癌患者仍未诊断出其癌性突变。
目前的突变研究几乎都是基于短读长的二代测序平台,长度在200-300bp(目前通用的是PE150bp),比较有利于检测点突变(snp和InDel )。虽然开发了一系列算法,但短读长测序仍然难以检测更复杂更大的结构变异,如染色体非整倍性、拷贝数变异CNV和染色体重排。
最近的长读长测序技术正在改变这种局面,比如单分子实时(SMRT)测序仪PacBio和Nanopore测序仪,纳米孔型测序仪MinION首次用于表征良性成人家族性肌阵挛性癫痫(BAFME)19号内含子中致病性重复序列扩张,作者以及他人的研究中也通过Nanopore测序方法检测到了癌症相关结构变异,另外Nanopore全长转录组测序甚至可以一条read可以揭示一个mRNA的完整剪接模式,并且Nanopore PromethION平台通量得到极大提升。
本研究作者试图用Nanopore测序技术研究肺腺癌中存在的之前未解析的致癌性突变。
研究方法
1)作者尝试使用PromethION平台对整个人类癌症基因组进行长读长测序。首先证明PromethION测序可以相对容易地识别点突变以及大的结构畸变和融合基因。并意外地发现,包含中小型结构变异的复杂组合突变非常普遍,构成了以前未定义的独特突变类别,称为致癌性局部拷贝数畸变(Cancerous Local Copy-number Lesions
,CLCLs)。
2)随后,利用长读长测序的优势,通过Nanopore全长转录组测序来表征全长转录本的结构。
3)最后,结合之前的其他多组学数据:基于Illumina短reads测序的全基因组重测序、RNA-seq和ChIP-seq,证明这些CLCL可能的功能,以及不仅限于细胞系中,也存在于临床样本中。
研究结果
1、癌细胞系长读长测序
使用纳米孔测序仪MinION及其更高通量PromethION平台进行了长读长全基因组测序分析。首先通过对LC2/ad的基因组进行测序来验证新型PromethION仪器的性能,LC2/ad是源自日本肺腺癌患者的肺癌细胞系。作为参考,作者从总共33个MinION runs(R9.5)中收集了全基因组测序数据,以31×的总测序深度覆盖了整个人类基因组,共有7,282,846条reads(93,813,338,154 base)。reads的*大长度和N50长度分别为2,495,160 bp和30,606 bp。采用Minimap2将67.5%的reads比对到人类参考基因组UCSC hg38。比对上的reads平均长度为16,452 bp,总体序列一致性平均为82%,比以前的长读长人类癌症基因组测序分析显著增长。PromethION测序需要大约三个flowcells才能产生总共10,064,668个reads(100,440,433,160 bp),总覆盖率为33x,Reads的*大长度和N50长度分别为987,834 bp和32,710 bp。使用Minimap2,将69.4%的读数定位到参考基因组。比对上的reads平均长度为13,620 bp,平均一致性为84.8%。值得注意的是,对于31X 93G数据量,PromethION平台不需要制备多个文库33次测序,因此与MinION相比,PromethION所需的起始DNA总量可减少十倍以上。
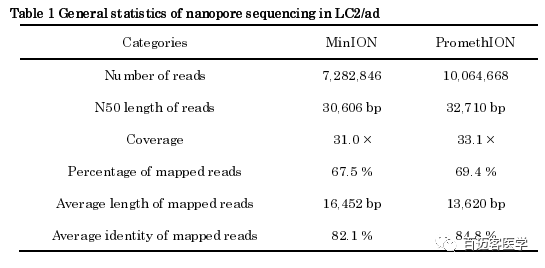
PromethION vs MinION:为了探究PromethION测序是否与MinION测序兼容,比较了获得的两个数据集的特征。Reads长度的总体分布是相似的(图1A)。这两个数据集都包含很大一部分超过50 kb的长读长reads(MinION:360,786个读,PromethION:451,698个),20X以上测序深度覆盖了超过50%的人类基因组区域(图1B),两个数据集均显示了超过80%的整体保真度(图1C),与先前的研究相似。PromethION平台各项指标稍微好一点。结论:PromethION是用于整个癌症基因组测序的有效分析方法。
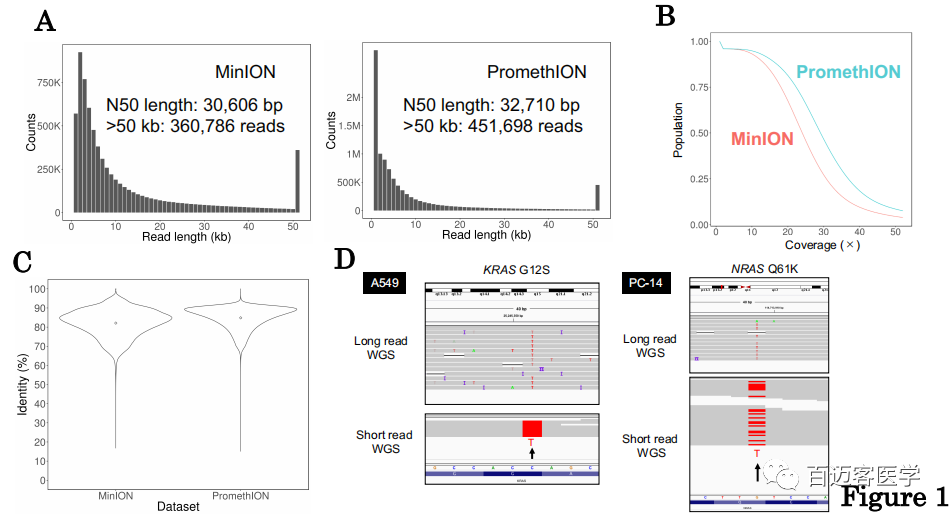
图1
在完成对从MinION和PromethION数据的初步评估后,作者对另外4个肺癌细胞系(A549、REFF-LC-KJ、RELF-LC-MS和PC-14)进行了MinION和PromethION测序,分别覆盖了9.5-18.5X基因组,例如,在RERF-LC-KJ测序中,产生了5,986,875个reads(总碱基数57,062,227,853 bp,18.5x),Reads*长长度和N50长度分别为922,768 bp和23,442 bp。
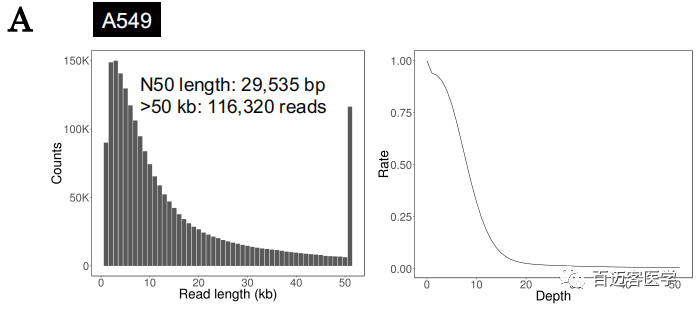
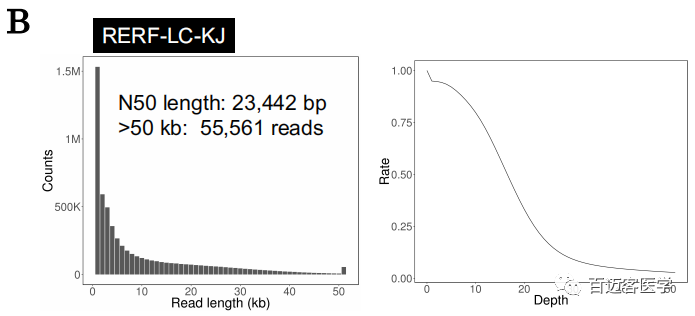
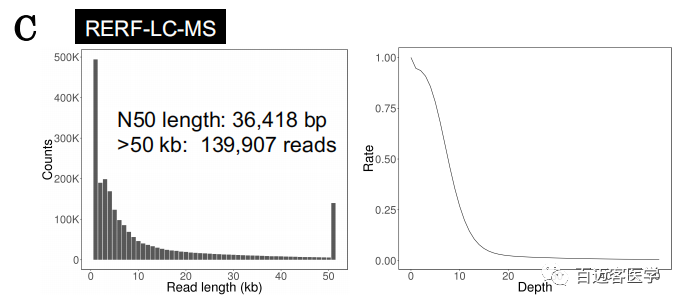
补充图1
为了评估单碱基水平数据质量,通过使用Integrative Genomics Viewer(IGV)手动检查了比对结果,针对相应细胞的已知驱动突变,在A549中,有11条reads显示出致癌突变KRAS G12S 点突变(左,图1D)。在PC-14中,8条reads显示驱动基因NRAS Q61K点突变(右,图1D)。REFR-LC-KJ和RERF-LC-MS细胞系中没有任何众所周知的驱动突变。所有这些结果与以前的报导一致。这些结果共同表明,至少当癌组织纯度(癌细胞含量)与培养细胞一样高时,仅使用长读测序也可以在单碱基水平上进行突变检测,即检测点突变。
2、大的基因组结构变异检测
使用长读长测序数据,尝试检测比点突变大的结构畸变(图2A)。从LC2/ad的MinION/PromethION数据集中,成功鉴定出12条reads与CCDC6-RET融合基因的junction位点直接重叠,这是该细胞系已知的“癌症驱动突变”(图2B)。进一步尝试鉴定大的缺失,前人报道抑癌基因CDKN2A基因周围有大的缺失,发生在LC2/ad,A549和PC-14细胞中。使用这项研究中的MinION/PromethION数据集,我们再次确认了该基因在各细胞中的缺失(图2C)。此外,每个CDKN2A缺失的√确连接点在细胞类型之间是不同的。
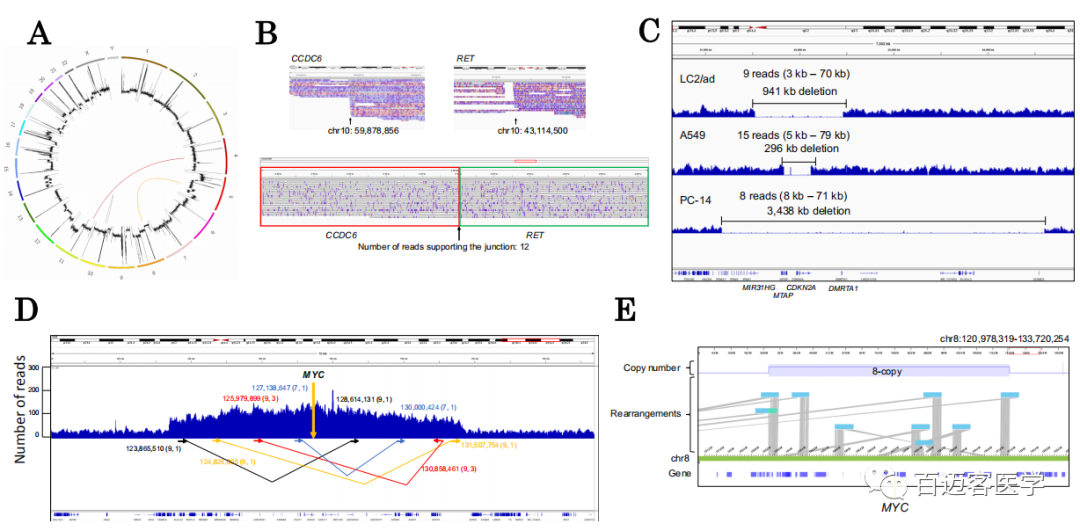
图2
通过采用split比对方法来检测新的融合基因(图2A),确定了3种新的重排,通过Illumina的短读长测序进一步证实了这些重排。LC2/ad中融合基因为NELL1-CCSER1和EFNA5-IKBKB,在RERF-LC-KJ中为UTS2B-GRM4融合基因。每种情况,长读长测序均能以单碱基分辨率√确鉴定连接点。
进一步尝试破译比较困难的MYC基因重排,确定了LC2/ad中MYC基因的拷贝数畸变。估计此扩增覆盖了中心为MYC基因的大约8Mb基因座。即使使用长读测序,仍然很难完全重建其结构,其中包括复杂的重排模式,在8号染色体上以估计的非整倍性为8扩展到8 Mb(图2D)。对于MYC区域,尝试通过光学图谱方法Bionano Saphyr识别正确的结构。即使使用Saphyr,MYC区域的√确结构仍然难以捉摸,尽管该分析的结果支持了跨越8 Mb区域的MYC扩增,具有大约8个拷贝(图2E)。
3、鉴定新的致癌结构变异CLCL
在尝试确定已建立类别的上述结构畸变(CNV、插入INS、缺失DEL、倒位INV、易位ITX/CTX)时,意外地发现了一种新型的局部结构变异(图3)。这些畸变由拷贝数改变、倒位和缺失的复杂组合组成,似乎不完全属于上述类别,因此将其命名为致癌性局部拷贝数畸变(Cancerous Local Copy-number Lesions,CLCL)。仅基于短读长reads很难识别和表征这些CLCL的√确连接,可能偶尔会获得一些提示性信息。
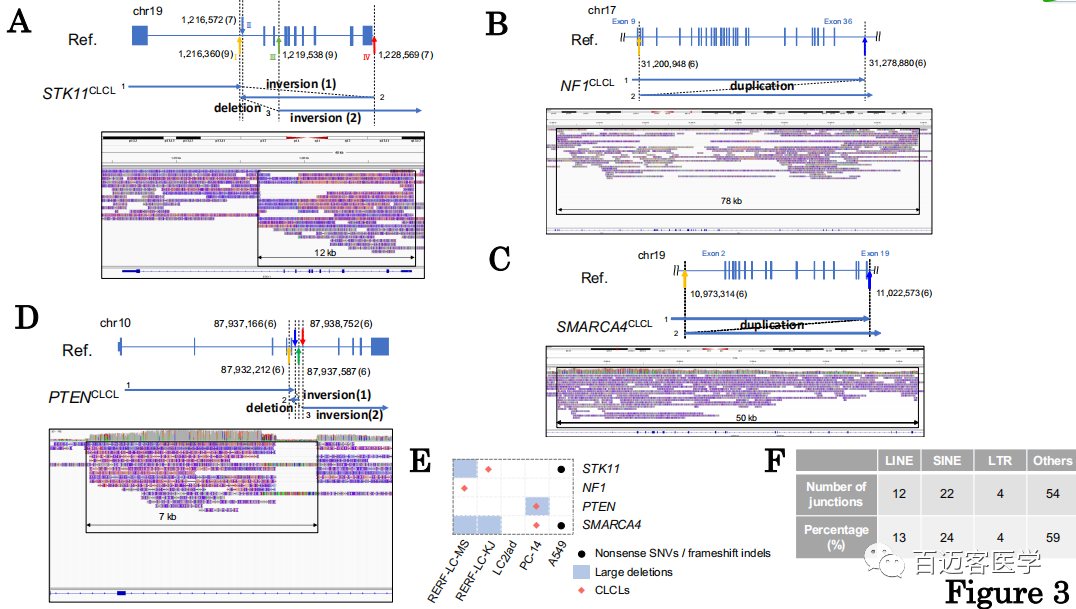
图3
第一个例子是在STK11基因座中发现的。在先前使用Illumina进行的肺癌全基因组测序研究中,发现RERF-LC-KJ细胞中STK11基因区域可能存在局部拷贝数变异,测序深度从内含子1的中间到基因的末端增加。存在短reads的split标签,这表明该区域可能发生反转。尽管在该区域定位了大量的测序reads,但仍无法重建其√确结构。
作者分析了长读长数据以破译STK11基因位点的畸变(图3A)。它揭示了如下畸变:第一次重排是从内含子1(chr19:1,216,572;断点II)开始,到基因下游(chr19:1,228,569;断点IV)。反向序列继续回到内含子1的中间(chr19:1,216,360;断点I,其是起始断点II上游的212个碱基),该序列返回并跳至内含子3(chr19:1,219,538;断点III)。后面的序列继续到基因座的末端。分别以7条和9条PromethION reads代表检测到的junction位点(断点II/IV和I/III)。当我们重新检查Illumina的reads时,在断点I和II之间以及断点III和IV之间的两个区域(图3A中的方框区域),测序深度都增加了。作者还使用软切割(soft-clipped)方法分析短读长数据发现,使用短读长split标签很难检测到两个断点I和III,部分原因是junction位点位于重复区域。
4、其他癌细胞系其他基因CLCL突变
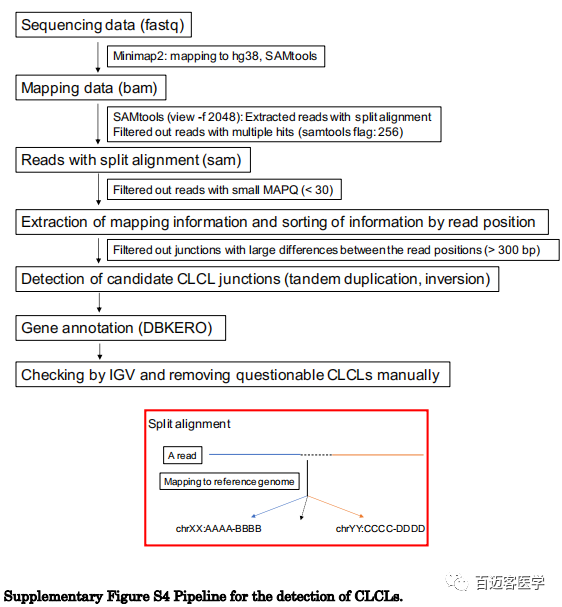
为了更普遍地识别所有肺癌细胞系其他基因座中的CLCL,作者构建了一条新的分析性生物信息学流程。简而言之,利用了来自比对结果的split比对信息,根据reads位置对比对信息进行排序,并提取了候选CLCL。相关的reads被重组以重建其结构。
在其他细胞系中也成功鉴定出以下数量的CLCL:LC2/ad中有16个,A549中有1个,RERF-LC-KJ中有7个,RELF-LC-MS中有7个,PC-14中有11个。重要的是,CLCLs甚至出现在关键的癌症基因中,例如STK11,NF1,SMARCA4和PTEN基因。异常的结构各不相同,并且由于其复杂的结构和受累区域的大小,传统的基于短读长测序分析的方法不易检测到它们中的大多数。相对简单的一种是在RERF-LC-MS细胞的NF1基因中检测到的(图3B),内含子9(chr17:31,200,948)和最后一个外显子36的下游区域(chr17:31,278,880;在交界处有6条reads支持)之间的串联串联重复。在另一种情况下,SMARCA4 CLCL的结构显示出从内含子1(chr19:10,973,314)到内含子20(chr19:11,022,573;在交界处有8条reads支持;图3C)。在PC-14中的PTEN结构中发现了更复杂的情况,该CLCL是倒位和缺失的组合(图3D)。在这些相对简单的情况下,将Illumina短reads重新映射到已发现的junction位点可验证重建结构的√确识别。
确实,尽管在先前的研究中部分怀疑了这些突变的存在,但在进行这项研究之前,它们的确切结构仍然难以捉摸。作者和其他人以前曾根据这些case的短读长测序数据,怀疑存在大的缺失、移码缺失和剪接位点突变。然而,通过基于短读长测序的常规变异检测,无法检测到某些情况,在本研究中,这些情况首先被确定为CLCL(图3E中黑点)。
作者还检查了CLCL的基因组背景。总计,有64%(28/44)的CLCL具有至少一个与长散布核元件(LINE)、短散布核元件(SINE)或长末端重复序列(LTR)重叠的junction位点,分别为13%、24%和4%(12/92、22/92和4/92)(图3F)。它们的独特位置可能会影响通过短读长测序对CLCL的√确识别。
5、与CLCL相关的异常转录事件
在很多细胞类型的许多关键基因中发现了新的CLCL型畸变后,立即提出的问题是它们以何种方式产生转录或表观基因组后果。全长转录组测序-使用MinION重新生成并分析了全长cDNA测序数据。并利用了以前的Illumina短读长RNA-seq和ChIP-seq数据。在RERF-LC-KJ细胞中,短读长序列数据表明STK11转录物在内含子1处异常剪接,并且转录跃迁到CLCL结构之前。代表全长转录本的MinION reads进一步指明√确的剪接模式和转录终止位点(图4A)。对于几乎所有的转录本,第一次剪接均发生在异常位置(来自chr19:1,216,268),并且转录根据CLCL结构发生(RNA-seq reads涵盖从chr19:1,216,572至chr19:1,228,569的断点II-IV)。在下游CLCL区域内也观察到一些异常转录(中间图,图4A)。在STK11基因是野生型的PC-14细胞中未观察到这种异常转录模式(下图,图4A)。
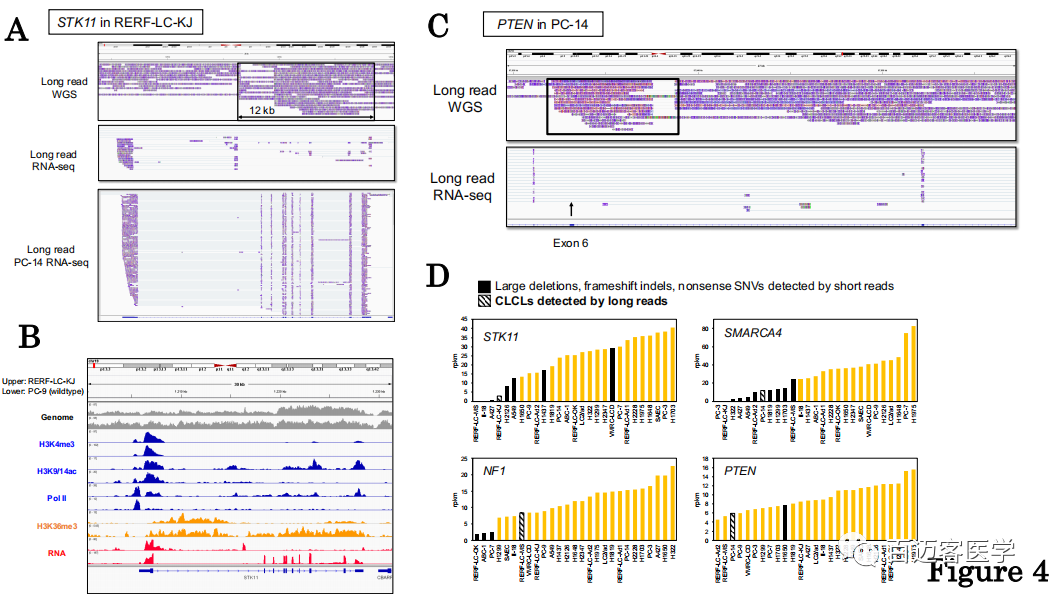
图4
作者检查了CLCL周围区域的表观基因组标记,以H3K4me3,H3K9/14ac和RNA聚合酶II的ChIP-seq表示。不管是否为携带CLCL或野生型STK11基因座细胞系中,染色质通常在启动子区域形成活性结构,并且转录通常在正确的位置开始(图4B)。然而,仅在具有CLCL的REFR-LC-KJ细胞中,H3K36me3标记在内含子1的中间消失,表明转录延伸应恰好在CLCL开始的地方被破坏。Illumina RNA-seq数据还支持RNA在内含子1的中间异常剪接,并根据CLCL结构进行转录。这些异常转录本的表达水平测得为2.8 rpkm。没有检测到正常的转录本。然而,尽管在某种程度上异常的转录本表达低于野生型,但仍保持了较高的表达水平。
我们对其他CLCL进行了类似的分析,对于PC-14中的PTEN基因(图4C),CLCL位于外显子6。结果,该外显子完全跳跃。因此,所得的转录本应移码,可能导致PTEN基因的功能丧失。根据Illumina RNA-seq数据检查了包含CLCL的STK11,NF1,SMARCA4和PTEN基因中的RNA表达水平。结果表明,CLCL通常可能导致基因表达水平降低(图4D)。但是,在某些情况下,基因表达水平仍然很显著,例如RERF-LC-MS细胞中的NF1转录本和PC-14细胞中的PTEN转录本。
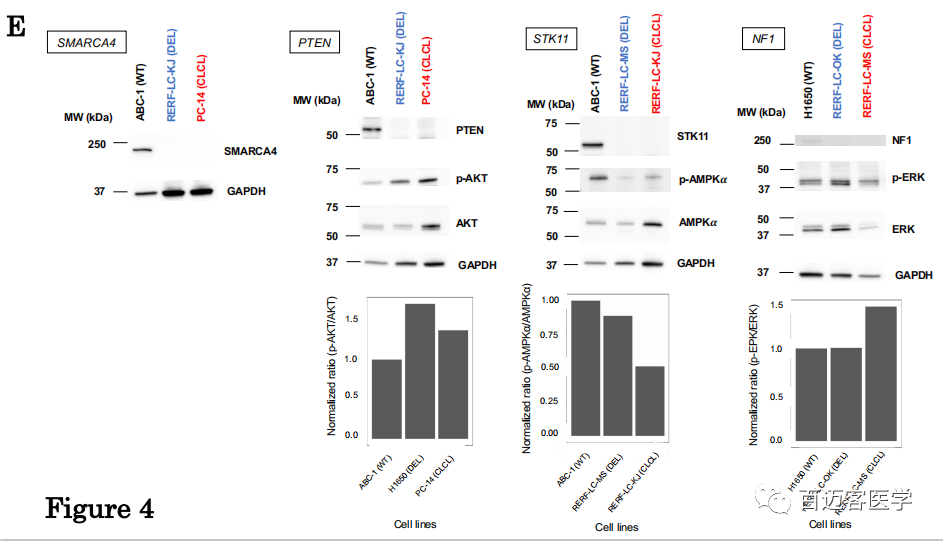
图4E
为了解决CLCL的生物学意义,作者研究了受CLCL影响的基因座如何引起蛋白质表达水平及其相关信号通路变化。蛋白质印迹WB分析发现STK11,NF1,SMARCA4和PTEN的蛋白质在这些基因中带有CLCL的细胞中完全丢失了(图4E)。进一步检查了下游蛋白的激活状态,在所有检查的case中均观察到了预期的信号通路破坏。磷酸化的AKT(phospho-AKT)相应地激活mTOR信号通路,而PTEN抑制AKT的磷酸化。观察到phospho-AKT异常上调,反映了PC-14细胞(PTEN-CLCL)中PTEN的功能丧失。AMPK是在维持细胞动态平衡中起重要作用的基因,AMPK蛋白在其α亚基处的磷酸化被STK11激活。在RERF-LC-KJ细胞(STK11-CLCL)中,其激活受到损害。NF1基因是RAS的负调控因子,位于RAS信号通路下游的磷酸化ERK在RERF-LC-MS细胞(NF1-CLCL)中异常上调。无论是由于常规畸变还是CLCL,相应基因的蛋白质明显丢失,但其后果仍会根据情况而有所不同。例如,即使STK11蛋白在RERF-LC-MS细胞(STK11缺失)和RERF-LC-KJ细胞(STK11-CLCL)中类似地消失,在RERF-LC-KJ细胞(STK11-CLCL)中磷酸-AMPKα的增强比例也更高。RERF-LC-OK(NF1-缺失)细胞系中NF1蛋白影响几乎无法检测到,而对RERF-LC-MS细胞(NF1-CLCL)的影响却很明显。表明,其他途径有时可以弥补关键蛋白的缺失。
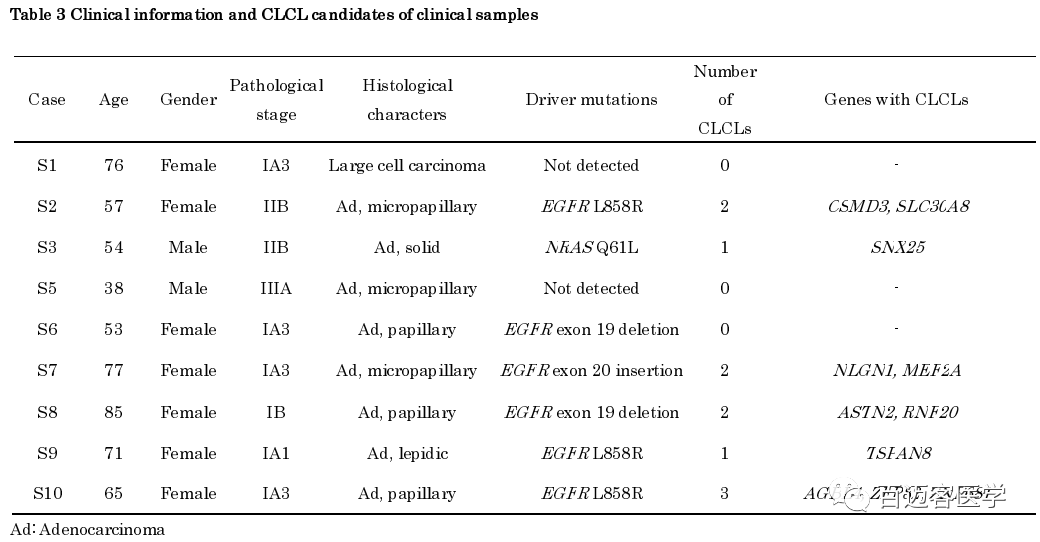
6、临床肺癌标本中CLCL突变检测
为了检查临床肺癌肺腺癌病例中是否也存在CLCL,对9名日本肺腺癌患者的手术标本进行了类似的PromethION全基因组测序(表3)。表3中显示了每个患者所检测到的驱动基因突变。平均每个case生成了43,953,136,203 bp的序列(深度大于10倍)。对于S10病例,还对正常组织标本进行了测序,以消除可能的正常组织变异和源自比对错误的可疑CLCL。
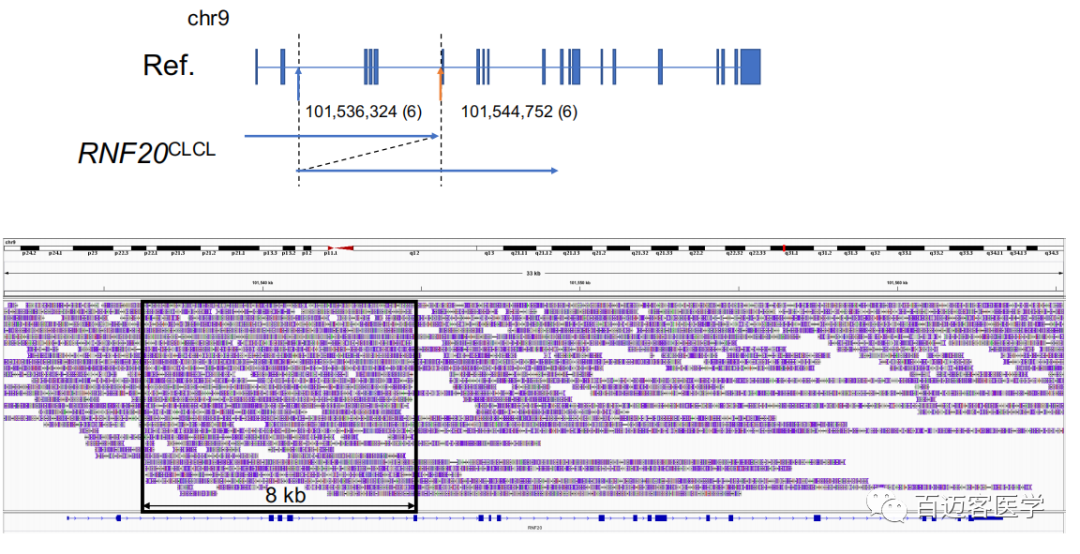
再次成功检测到CLCL,9个样本中有6个在其肿瘤基因组中至少包含一个CLCL,包括几个关键的癌症基因。例如,在病例S8中,确定了RNF20 CLCL。该患者为女性患者,已被证明具有EGFR外显子19缺失作为驱动突变。但是,其他癌症突变仍然难以捉摸。RNF20基因的CLCL为内含子2(chr9:101,536,324)和内含子6(chr9:101,544,752)之间的串联重复,这很可能导致该基因的功能丧失。RNF20基因编码具有抑癌功能的E3泛素连接酶,并且经常发生突变,特别是在肺癌中。表3总结了获得患者致癌作用的分子病因学指征。需要进一步扩展长读长测序序列,以更√确地识别CLCL的频率和CLCL偏好的基因。
7、根据公共短读长测序数据重新评估可能的CLCL
作者尝试利用预先存在的Illumina短读长测序数据分析CLCL,希望即使从短读长测序数据中也能够识别出候选CLCL。并且作者对先前的短读长reads如何展示这些CLCL感兴趣。
为了从短读长序列识别CLCL,采用软切割程序GenomonSV(https://github.com/Genomon-Project/GenomonSV)。选择“split” reads作为“软切割”reads,以及成对末端reads,它们可能跨越SV的junction位点。作为模型数据集,首先分析了用于上述PromethION测序的5种肺癌细胞系的全基因组短读长测序数据。对于每个细胞系,在基因区域平均提取182个“soft-clipped”连接点。将串联重复结构定义为短读长测序数据中的假定CLCL。在细胞系中平均有26个基因受到推定的CLCL的影响。比较从短读长和长读长中检测到的CLCL,在PromethION检测到的CLCL中,从短读长序列数据中也检测到72%的基因(图5A)。但是,由于假阳性检出率普遍较高,因此z确率限制为25%。
之后收集并分析了9个临床病例约63X测序深度的全基因组短读长测序数据,分析可能的CLCL,确定了可能受推定的CLCLs影响的9个基因。如图5B所示,从短读长数据开始检测到CLCL,估计灵敏度为73%。但是,由于较短的读长reads所固有的各种原因,z确率被限制为14%。
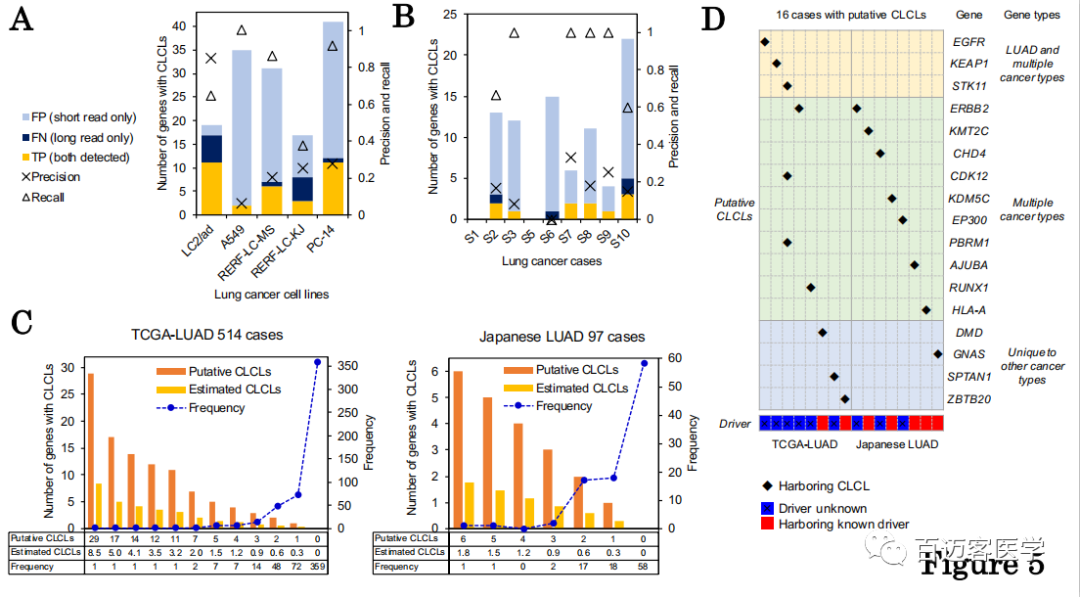
图5
尽管对于所有细胞系和临床样品,使用短读长数据分别估计21%和72%的CLCL检测z确度和重复检出率,但将构建的分析流程应用于514个 TCGA肺腺癌(TCGA-LUAD)和97个日本肺腺癌(Japanese LUAD)样本全外显子组测序数据,通过分别从TCGA-LUAD和日本LUAD病例中进行软切割reads分析,共检测到269个和50个具有串联重复结构的连接点,这些结构可能对应于CLCL(每个病例1至29个基因)。总计从155(30%)TCGA-LUAD和39(40%)日本LUAD病例中提取了候选CLCL(图5C)。
接下来作者分析了这些病例中是否携带299个与癌症*相关的基因CLCL,检测到16例(514 + 97例中为2.6%),在17个癌基因中具有潜在的CLCL(图5D)。这些病例中有9个没有已知的驱动基因突变。例如,TCGA-49-4512(女性,非吸烟者),在EGFR基因的激酶结构域中鉴定出潜在的CLCL。先前曾报道过这种重复,可能会导致EGFR异常激活,从而成为该病例的驱动突变,应通过afatinib/阿法替尼等EGFR抑制剂解决该患者的治疗目标。在另外2个病例(男性和吸烟)中检测到与ERBB2相关的假定CLCL,似乎在ERBB2基因区和下游基因间或基因区之间出现了异常重复。其他患者在其他重要的抑癌基因(如STK11和PBRM1)中也带有假定的CLCL,其突变状态可用作免疫检查点抑制剂的假定标记物。对于这些情况,尚不清楚推定的CLCL的√确结构以及功能相关性,因此,应对它们进行详细的长读长测序分析。
小结
本研究中描述了使用PromethION在肺癌基因组中识别和表征结构畸变,揭示了由局部重复、倒位和微缺失的复杂组合组成的独特结构畸变CLCL,进一步分析并发现,即使在关键的癌症相关基因中,这些突变也发生在体内,这些突变可能阐明了致癌性事件和治疗策略仍然难以捉摸的患者的分子病因。这是将PromethION测序用于癌症基因组学的研究。显然,需要对测序方法本身进行进一步的改进,并对计算方法进行改进,以达到进一步的目标。确实,这项研究提出的问题多于答案。从这个意义上讲,这仅仅是首次研究,为更全面地了解癌症的复杂基因组畸变以及进一步深入研究其生物学铺平了道路。




 京公网安备 11011302003368号
京公网安备 11011302003368号