
分类: 医学研究
1、 安装软件

pyGenomeTracks使用conda、pip和源码安装,推荐使用conda安装
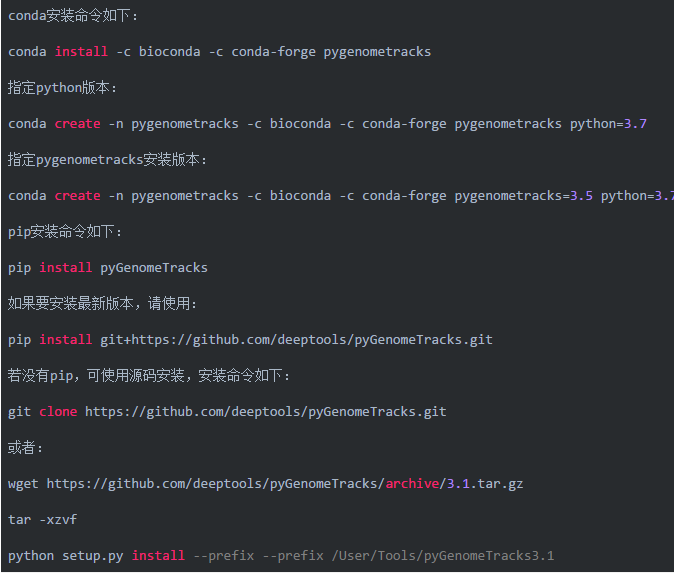
参考网址:https://pygenometracks.readthedocs.io/en/latest/content/installation.html
2、 绘图文件
pyGenomeTracks旨在产生高度可定制的高质量基因组浏览器轨道。目前,可以绘制:

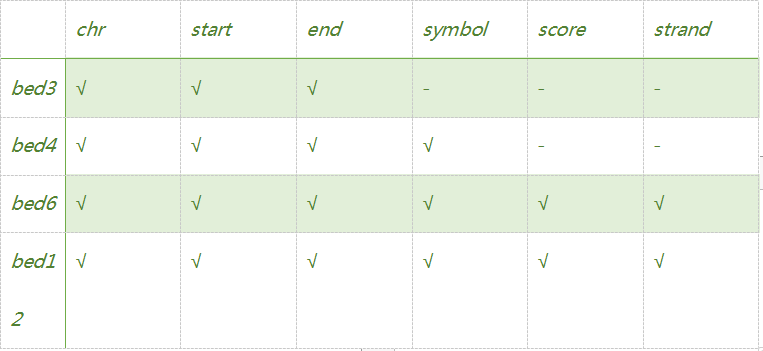
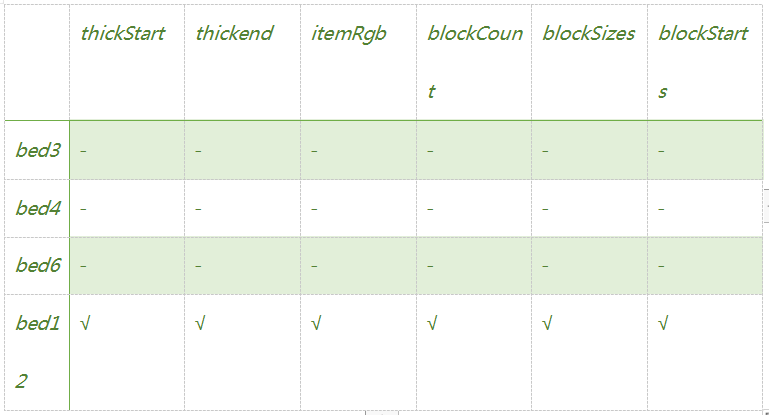
具体格式说明,见
https://www.jianshu.com/p/4880f1969919

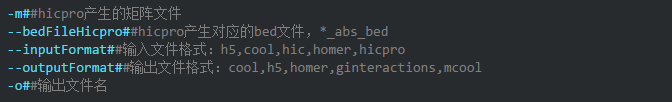
3、 使用过程
第一步:准备绘图配置文件:可自动生成,也可以按照模板手动填写
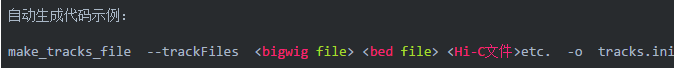
参数说明:
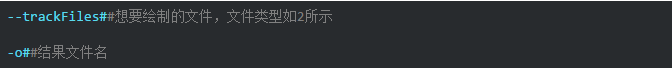
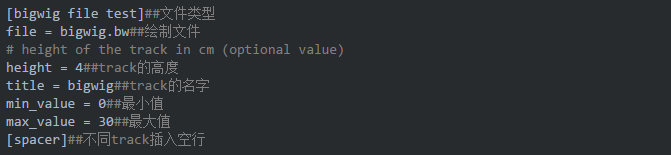
单个track
![]()
生成绘图配置文件tracks.ini如下:
第二步:绘图代码
![]()
参数说明:
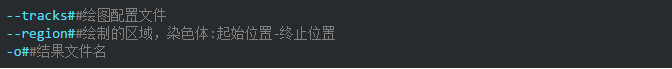
生成的结果如下:
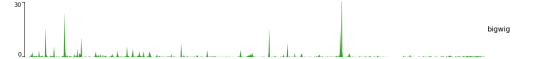
多个track
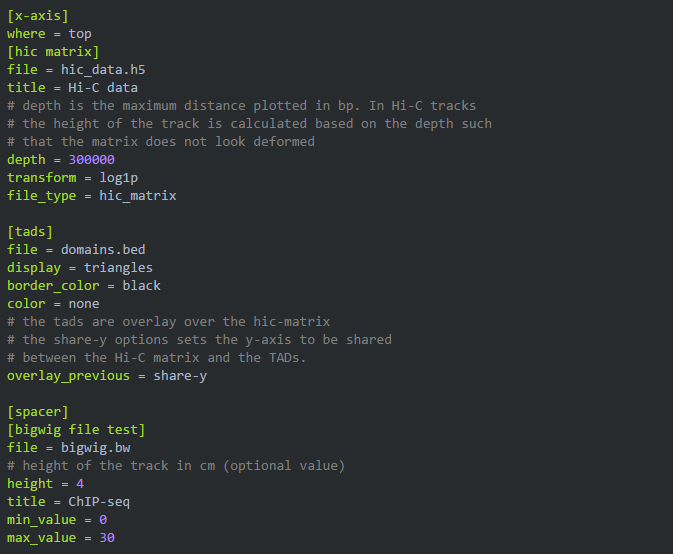
结果如下:
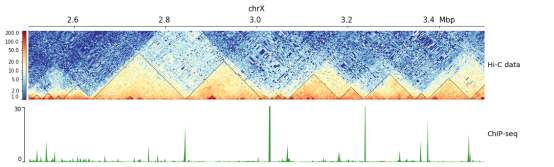
pyGenomeTracks还可以绘制更复杂的图形哦!
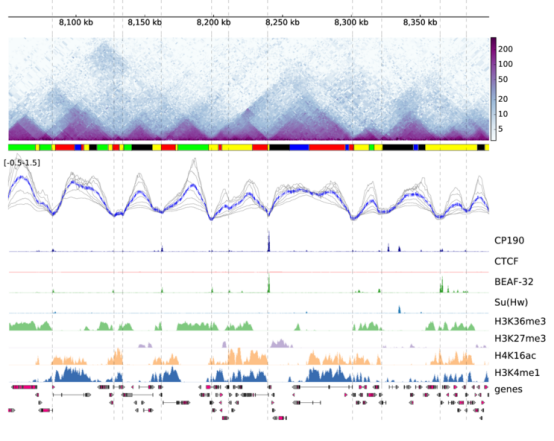
多组学分析的时候经常会对相同的文件绘制不同的区域,pyGenomeTracks可生产一个region.bed文件,进而对相同的文件绘制不同的区域:

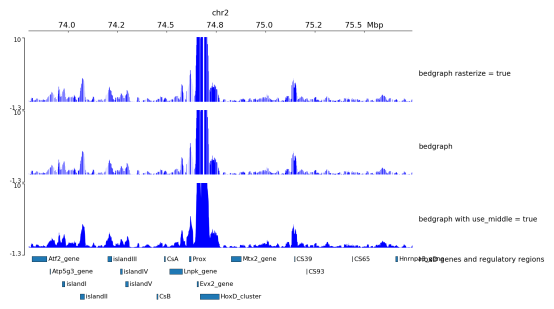
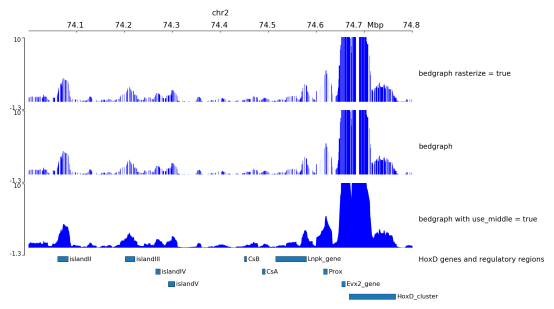
更多示例见:https://pygenometracks.readthedocs.io/en/latest/content/examples.html
最近文章



 京公网安备 11011302003368号
京公网安备 11011302003368号