
随着对肿瘤细胞的糖代谢重编程的认识,有氧糖酵解对肿瘤治疗的抑制作用越来越受到重视。糖酵解通路中的许多关键蛋白已被发现是治疗和克服耐药性的重要靶点。有氧糖酵解抑制作用的研究主要集中在实体肿瘤来源的细胞模型中,而白血病细胞模型中信息较少,尤其是白血病多药耐药(MDR)细胞。因此,有必要从肿瘤代谢的角度研究MDR的分子机制,探讨白血病MDR细胞中有氧糖酵解对糖代谢和药物敏感性的影响。
12月新鲜出炉的公司合作文章中,兰州大学基础医学院甘肃省新药临床前研究重点实验室的研究者借助转录组测序技术,探究了白血病MDR细胞的代谢重编程机制,对于发现通过恢复白血病MDR细胞药物敏感性以改善治疗效果的治疗策略有重要意义。
这篇文章中,作者在得到转录组测序筛选到的差异表达基因后,集中于“糖代谢”这一主要研究目的,通过差异表达基因在功能和通路上的注释及富集分析,找到了关键的糖代谢相关基因,以及药敏细胞和耐药细胞间发生变化的信号通路,为后续研究奠定了坚实的基础。这也是一种高效的转录组测序数据深入挖掘方法。接下来,小编跟大家一起看看这篇文章吧。
研究背景
葡萄糖是癌细胞中最丰富的能量来源之一。除了食物摄取以外,葡萄糖主要来源是糖异生和糖原分解,属于葡萄糖合成代谢途径。相反,线粒体氧化磷酸化(OXPHOS)和糖酵解是葡萄糖分解代谢的两条主要途径。
常氧的正常细胞中,高水平的OXPHOS是细胞ATP的主要来源。然而,近年来发现,尽管存在充足的氧气,但许多癌症仍明显依赖糖酵解作为一种重要的能量来源,即“有氧糖酵解”或“瓦博格效应”,这已被广泛认为是代谢重编程的共同特征。此外,有氧糖酵解可能赋予肿瘤细胞以耐药性,可能的原因有:1)细胞内微环境酸化,可能会降低药物吸收和效率;2)支持细胞增殖所需的大分子合成的中间代谢物增加;3)通过减少氧自由基来抗氧化损伤。此外,在化疗作用下,肿瘤/白血病细胞可能会表现出额外的代谢重编程,从而获得对抗肿瘤药物的耐药性。
阿霉素(ADM)是一种具有强烈毒性的抗肿瘤药,临床上用于治疗急性淋巴细胞白血病、急性粒细胞白血病等。前人研究和作者前期的研究表明,K562/ADM耐药细胞比其亲代K562敏感细胞具有更高的有氧糖酵解活性,这意味着耐药白血病细胞通过糖酵解重编程获得了对细胞毒性药物的适应性。
在此,作者采用二代高通量测序技术对两个细胞系间的差异表达基因(DEG)进行分析,高度富集的DEG则为候选耐药性相关基因。糖酵解途径中,乳酸脱氢酶(LDHA)催化丙酮酸生成乳酸是关键步骤,而草酸是一种LDHA抑制剂,可用于抑制有氧糖酵解。用草酸和ADM单独或联合处理后,测定其对细胞活力、ADM细胞毒性、糖代谢和候选耐药相关DEG表达的影响。
研究方法
1、材料
药敏细胞系–K562人类白血病细胞系,ADM耐药细胞系–K562/ADM细胞系。
2、转录组测序
两种细胞系各三个样本,提取总RNA,构建转录组文库,进行转录组测序。
3、体外细胞毒性试验
4、糖代谢指数测定试验
5、Western blot
研究结果
1、转录组测序发现与糖代谢相关的基因
K562/ADM细胞与其亲本K562细胞间基因表达谱的比较将为MDR的研究提供有价值的机制见解。药敏细胞和耐药细胞的基因表达比较显示,两者转录组之间存在巨大差异,共检测到1742个DEG。然后,通过与GO、NR、Swiss-Prot、eggNOG和KEGG数据库的比对,鉴定到了205个与代谢相关的DEG。其中,相比于药敏细胞,耐药细胞中有97个基因表达上调、108个基因表达下调,表明这些基因可能在K562/ADM细胞的耐药性中起重要的作用。
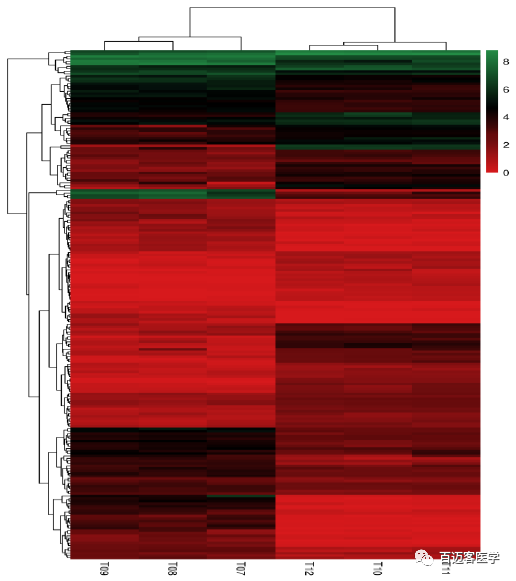
DEG表达量聚类热图
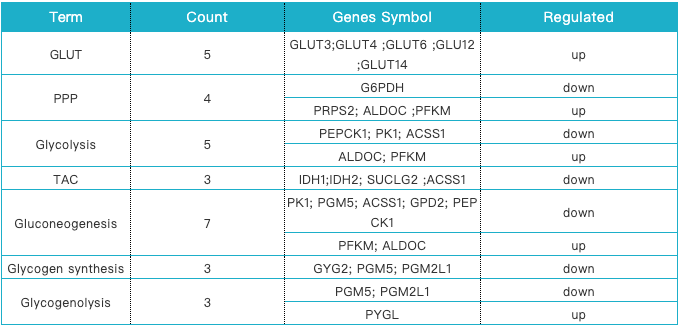
作者进一步鉴定了葡萄糖代谢中的酶和转运体的相关DEG,发现K562/ADM细胞中GLUT3和GLUT4的基因表达水平较高,这些基因作为葡萄糖转运体促进葡萄糖的摄取。K562/ADM 细胞中,编码糖酵解代谢相关酶(PFKM和ALDOC)的基因表达上调,而参与TCA的DH1、IDH2和SUCLG2基因则表达下调。此外在耐药细胞中,糖异生关键酶基因PEPCK1表达下调,糖原分解关键酶基因PYGL表达上调,而参与糖原合成的关键蛋白基因GYG2表达下调。有趣的是,与前人的蛋白组学研究结果一致,耐药细胞中PPP通路的关键酶G6PD下调。然而,参与嘌呤和胸腺嘧啶合成的关键酶PRPS2上调。
结果表明,白血病MDR细胞表现为糖代谢重编程。事实上,在前人和作者之前的工作中,也观察到与药敏细胞相比,耐药细胞的糖酵解通量有所增加。
表1 糖代谢相关DEG
此外,KEGG聚类和富集分析显示,与K562细胞相比,K562/ADM细胞中存在多个生物学过程的变化。为了进一步阐明糖代谢重编程的分子机制,将DEG比对到参与葡萄糖代谢的相关信号转导途径。所有DEG都是比对到了信号转导途径,如MAPK、PI3K-AKT、HIF、Ras等。如图,PI3K-AKT通路是DEG富集最多的通路。PI3K-AKT通路通过调节代谢分子(包括LDHA、HK-II和GLUT)的表达或移位,作为代谢稳态的关键调控因子。结果显示,PI3K-AKT通路是K562/ADM细胞中最富集的上调通路,与糖代谢重编程和MDR的发展有关。
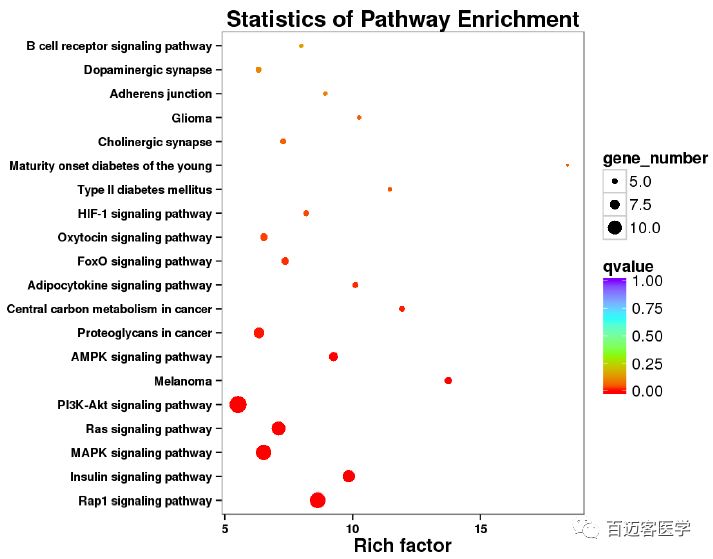
葡萄糖代谢的相关信号转导途径
2、草酸可以有效恢复K562/ADM细胞对ADM的敏感性
如图,单独用草酸处理的两个细胞系均呈现时间和剂量依赖性细胞毒性,但是细胞毒性相对较低。相比于单独用ADM处理,用ADM和草酸联合处理后,K562细胞和K562/ADM细胞增殖抑制率分别提高了为2.28±0.56倍和6.08±0.77倍,K562/ADM细胞增殖抑制率的增加倍数明显高于K562细胞。
这些数据表明,尽管耐药的K562/ADM细胞系对药物的敏感性不如其亲本细胞系,但与草酸联合使用时,其疗效显著提高。因此,抑制糖酵解可使K562/ADM细胞恢复ADM敏感性。
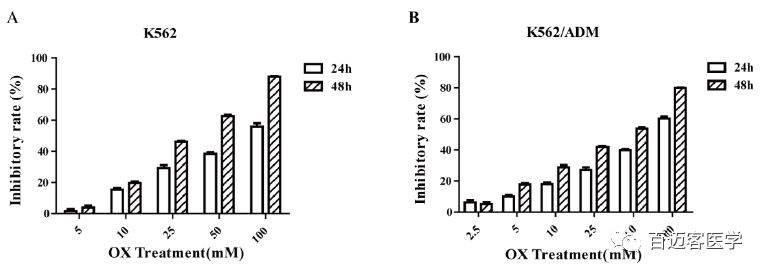
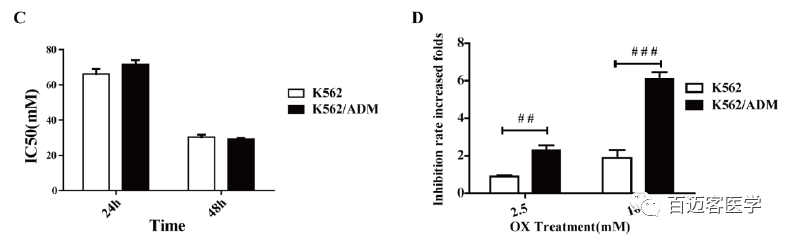
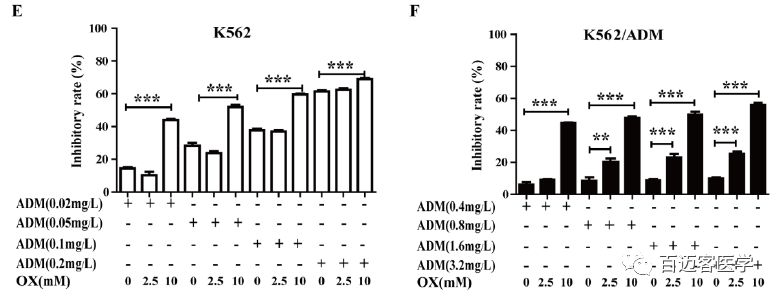
草酸和ADM对药敏、耐药细胞的影响
3、草酸对糖代谢通量和LDHA活性的抑制作用
在上述试验中,作者发现ADM联合草酸处理增加了人白血病细胞的细胞毒性。然后,作者研究了草酸对ADM处理的K562和K562/ADM细胞系糖代谢作用的影响,发现相比于ADM单独处理,ADM和草酸共同处理48h后,两个细胞系的葡萄糖消耗和乳酸生成的抑制作用均有所增强。草酸导致两种细胞系糖酵解抑制呈现剂量依赖性增加,而耐药细胞糖酵解抑制的变化倍数更高,这与联合处理时耐药细胞疗效升高倍数更高所一致,说明草酸对糖酵解的抑制导致葡萄糖消耗和乳酸生成的减少,导致白血病细胞对ADM的敏感性增加。
相对LDHA活性检测结果表明,草酸对K562细胞LDHA活性的抑制作用高于K562/ADM细胞。说明在K562细胞和K562/ADM细胞中,草酸对糖酵解通量的限制作用是不同的,不能用只通过竞争性抑制LDHA活性来解释,因此草酸抑制LDHA并不是提高ADM疗效的唯一机制。
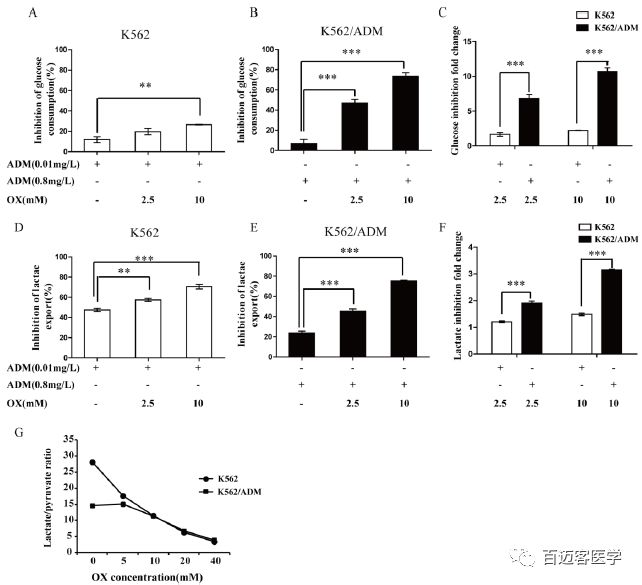
4、草酸抑制ADM诱导的AKT-mTOR-c-Myc通路的过度激活
上述转录组测序结果显示,在K562/ADM细胞中,PIP3-AKT通路上调,此通路是糖代谢稳态的关键调控因子,可能与耐药性增强有关。此外,草酸对糖酵解的抑制有效提高了K562/ADM细胞的ADM细胞毒性。此外,作者推测草酸诱导的LDHA抑制并不是草酸提高ADM疗效的唯一机制。因此,为了研究草酸的使用是否也影响了PIP3-AKT通路,作者用草酸、ADM单独或联合处理K562细胞和K562/ADM细胞后,通过western blot检测PIP3-AKT通路中一系列重要信号分子的表达水平。
结果表明,草酸逆转了ADM诱导的K562/ADM细胞中t-AKT和p-AKT473的上调、t-mTOR和p-mTOR的上调。在K562/ADM细胞中,ADM和草酸联合处理时显著下调的AKT-mTOR-HIFα可能导致更有效地抑制AKT-mTOR-c-Myc的过度活化,从而影响ADM耐药性。而AKT/mTOR/HIF/c- Myc的下游效应物—GLUT4和HK-II检测结果表明,ADM和草酸联合处理后,K562/ADM细胞中的两种效应物变化趋势与mTOR相似。
因此,cADM和草酸联合处理不仅可以直接竞争LDHA活性,也会中和耐药细胞中ADM引起的AKT-mTOR-c-Myc通路超活化,以及进一步的AKT-mTOR-HIFα-GLUT4/HK-II受损。
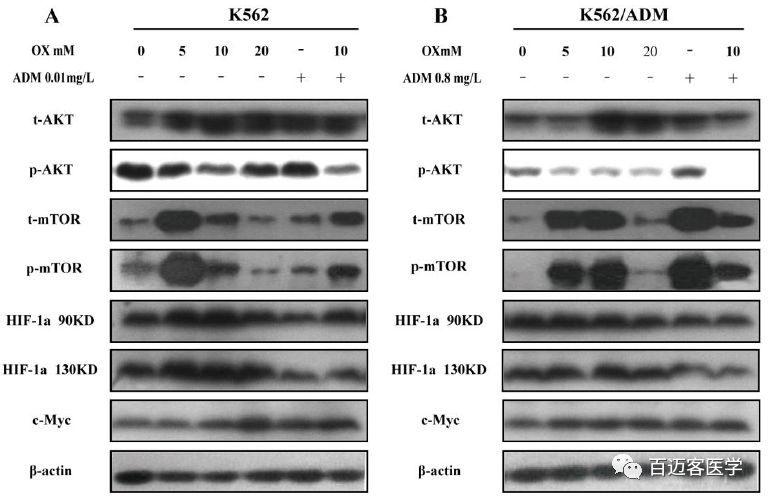
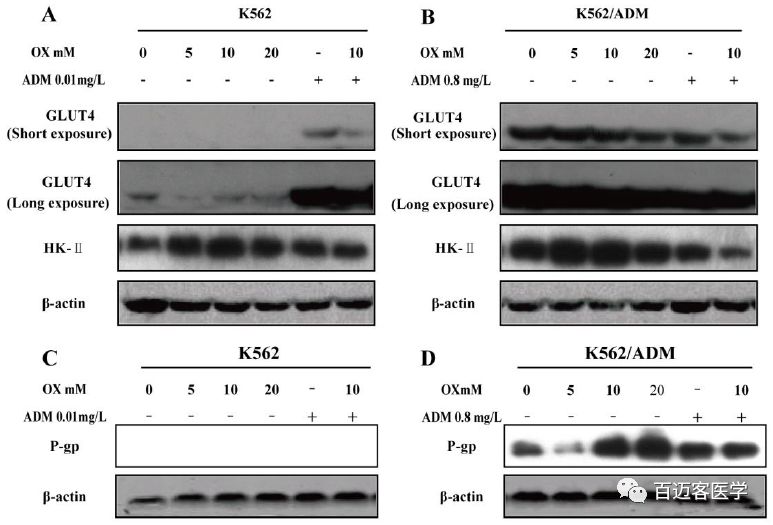
草酸和ADM对PIP3-AKT通路的影响
总结
为了研究有氧糖酵解在白血病耐药中的分子机制和作用,作者通过转录组测序发现了MDR白血病细胞系K562/ADM与其亲本-药敏细胞系K562的差异表达基因,并对DEG进行聚类和富集分析。采用乳酸脱氢酶抑制剂–草酸评价糖酵解抑制对K562/ADM细胞ADM敏感性及关键DEG表达的影响。
作者发现K562/ADM细胞与K562细胞共有1742个DEG。参与糖代谢的单基因编码酶的差异表达基因表明,K562/ADM细胞的有氧糖酵解通量较大,而PI3K-AKT信号通路与糖代谢有关,在K562/ADM细胞中表现出明显上调。草酸通过下调AKT-mTOR通路直接或间接抑制有氧糖酵解,改善ADM在ADM耐药细胞中的治疗效果,并使其重新敏化。
总之,ADM耐药性是由有氧糖酵解的增加介导的,与MDR白血病细胞中AKT-mTOR-c-Myc通路的过度激活有关。抑制有氧糖酵解和下调有氧糖酵解中的相关信号通路是一种潜在的使得白血病细胞致敏,从而克服MDR的化疗策略。
参考文献
Zhang X , Chen J , Ai Z , et al. Targeting glycometabolic reprogramming to restore the sensitivity of leukemia drug-resistant K562/ADM cells to adriamycin[J]. Life Sciences, 2018.




 京公网安备 11011302003368号
京公网安备 11011302003368号