

2019年6月14日,美国罗斯威尔帕克癌症研究所的刘涛等人在NAR(Nucleic Acids Research,核酸研究)杂志上发表了针对ATAC-seq开发的分析工具。

ATAC-seq(Assay for Transposase-Accessible Chromatin with highthroughput sequencing),基于高通量测序的染色质转座酶可接近性实验。在核小体连接致密的地方,转座酶不能进入,而松散的区域,转座酶能够进入并切割下暴露的DNA区域,并同时连接上特异性的接头,有接头的DNA片段被分离出来,用于高通量测序。ATAC-seq已被广泛用于鉴定基因组中可接近的染色质区域。 然而,目前的数据分析仍然使用最初为ChIP-seq或DNase-seq设计的方法,而没有考虑含有额外核小体定位信息的转座酶消化的DNA片段。
因此,作者提出了针对ATAC-seq数据的分析工具,HMMRATAC,一种基于半监督机器学习方法。 HMMRATAC软件的主要思想是decomposition and integration(分解和集成),首先将单个ATAC-seq数据集分成无核小体(nucleosome-free regions, NFR)和核小体富集信号,用隐马尔科夫模型(HMM)训练开放区域周围独特的染色质结构,然后预测整个基因组中的开放区域。 该方法利用了ATAC-seq数据的独特特征来识别的染色质的结构更准确。
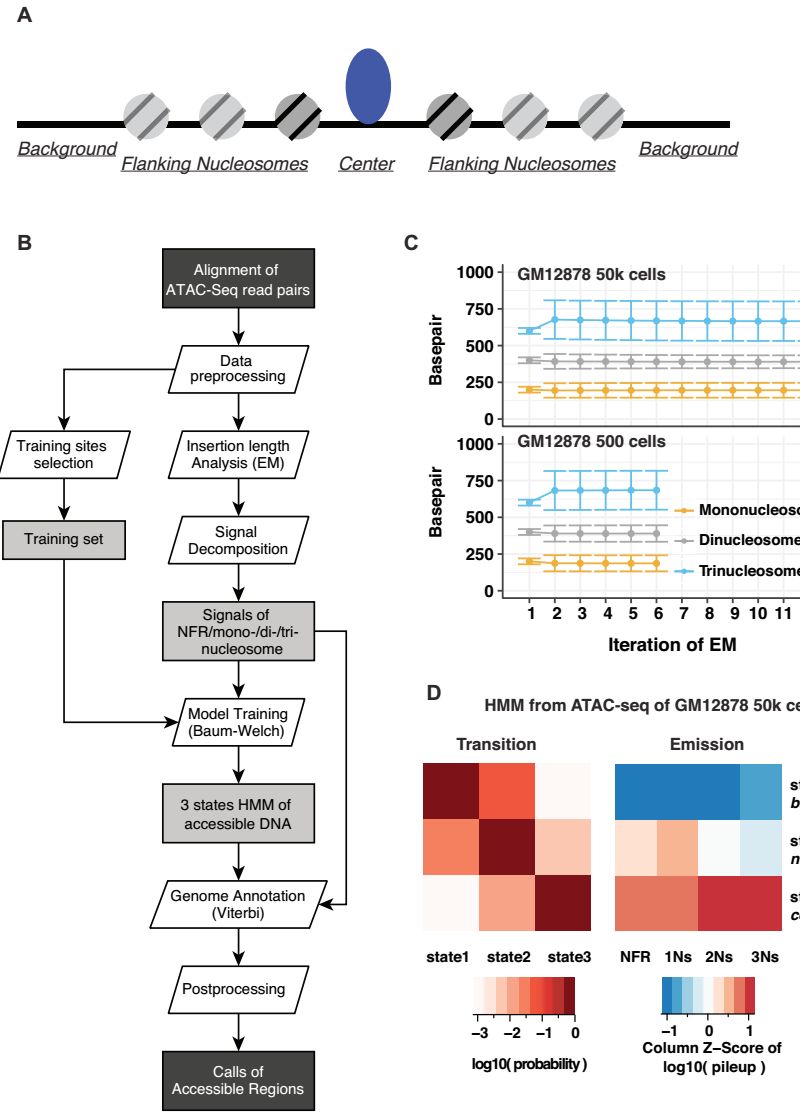
将HMMRATAC软件应用于已发布的人类ATAC-seq数据集上,发现其结果优于其他流行峰值调用算法。 同时作者还发现,相比于未经片段大小选择的配对末端数据,单端测序或片段选择的ATAC-seq数据集会导致灵敏度降低。
结束语
偷偷的告诉你,这篇文章的作者也是当年风靡一时现在还如日中天的MACS软件的作者呦!而MACS2,MACS14都是当前用于CHIP-seq和ATAC-seq中识别Peak区域用的最多的软件。
参考文献:
Evan D Tarbell, Tao Liu. HMMRATAC: a Hidden Markov ModeleR for ATAC-seq. Nucleic Acids Research, 2019.
Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. doi:10.1186/gb-2008-9-9-r137.
【相关阅读】
前沿|ATAC-SEQ专题-GENOME BIOLOGY & GENOME RESEARCH



 京公网安备 11011302003368号
京公网安备 11011302003368号