
2019年新年伊始,百迈客与中山大学中山医学院合作发表了RNA-seq & ATAC-seq多组学文章,研究了人类免疫缺陷病毒1型(HIV-1)潜伏感染的分子机制,文章发表在Elife杂志上,详情如下:
英文标题:TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb
发表杂志:Elife
影响因子:7.616
合作单位:中山大学中山医学院
发表时间:2019.01.17
研究背景
HIV-1潜伏感染是利用抑制性联合抗逆转录病毒治疗(combined antiretroviral therapy,cART)后实现治愈的一个主要障碍,其在体内存在病毒储藏库,需要终身的联合抗逆转录病毒治疗。潜伏感染的静息CD4+ T细胞不能产生足以被免疫系统识别的病毒抗原,因此很难被根除。作为功能性治疗策略之一的“shock and kill”策略已经引入并在这些年中得到广泛应用。然而,一些感染细胞含有非诱导型前病毒,其很难被LRA重新激活。进一步阐明HIV-1潜伏期的机制将有助于我们更好地了解病毒储藏库的形成和维持,并开发新的治疗干预措施。
(“Shock and kill”即“先激活后绝杀”策略,是利用药物激活潜伏在T细胞内的艾滋病毒使其暴露,随后通过增强免疫系统或者抗病毒药物消灭携带有病毒的宿主细胞。其中,“Shock”策略中很重要的就是潜伏期逆转剂(Latency reversing agents,LRA)的研发;“kill”即对激活后的HIV病毒进行杀伤,用的是用自体过继免疫疗法,如CTL反应和抗体依赖性细胞介导的细胞毒性作用(antibody-dependent cell-mediated cytotoxicity,ADCC)。)
表观遗传调控有助于建立和维持HIV-1潜伏感染:比如组蛋白脱乙酰酶、组蛋白甲基转移酶等”writer”在HIV-1长末端重复序列(LTR)上作抑制性表观遗传标记,由”reader”蛋白蛋白HP1γ和L3MBTL1进一步维持;miR-28等microRNAs和NEAT1等lncRNA,这些非编码RNA能够靶向病毒RNA和病毒蛋白,以介导HIV潜伏感染的转录或转录后调控。
重新激活潜伏HIV-1的另一个障碍取决于转录控制:转录起始水平,HIV-1潜伏期由转录因子(包括NF-κB、Sp1、AP-1、NFAT和TFIIH)缺陷以及转录抑制因子(包括LSF、YY1和CTIP2)的积累所贡献。然而,细胞周期蛋白Cyclin T1的表达在潜伏感染的细胞中下调,细胞周期蛋白依赖性激酶 CDK9也是无活性的。
虽然许多工作揭示了HIV-1潜伏期的表观遗传和转录调控机制,但仍存在一些重要问题:1、可能有一个多功能因素负责这两种机制;2、启动子近端暂停的机制尚未完全阐明3、P-TEFb如何被适当隔离、释放并靶向HIV-1启动子;4、尚未找到能够有效重新激活潜伏HIV-1的强大逆转剂LRA。
研究方法
- siRNA文库高通量筛选:鉴定HIV-1抑制和潜伏的细胞靶蛋白,TZM-BL细胞系(一种携带有荧光素酶报告基因的传代细胞系,并且该报告基因受HIV-1 tat原件调控,只有在HIV-1 Tat蛋白存在的情况下才能有效表达。)
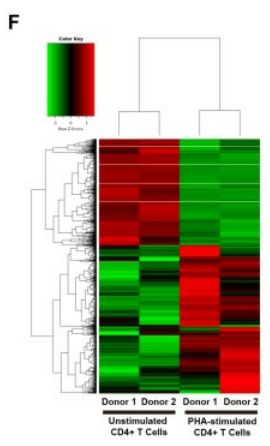
- RNA-seq:用PHA刺激来自两个健康供体的新鲜分离的CD4 + T细胞2天或不进行处理,鉴定HIV-1潜伏相关基因
- ChIP实验:TZM-bl和J-Lat 10.6细胞系鉴定TRIM28结合区域,以及后续各个组蛋白修饰或蛋白的chip鉴定
- SUMO-MS:TRIM28 SUMO化候选底物
- 蛋白功能阈缺失实验:探究TRIM28的功能及重要功能域
- 共定位实验:超分辨率的连续随机光学重建显微镜(cSTORM)3D共定位及结构光显微镜(SIM)共定位
- ATAC-seq:J-Lat 10.6或TZM-bl细胞系,研究TRIM28缺失后HIV-1启动子的染色质可及性
- 赖氨酸突变实验及逆转突变实验以及SUMO-MS等:鉴定特异性SUMO化位点
- 细胞毒性相关实验:细胞毒性测定、细胞活性测定、细胞数计数和细胞增殖测定
- 等
研究结果
1、TRIM28抑制HIV-1表达并导致HIV-1潜伏期
为了确定可能导致HIV-1抑制和潜伏的细胞靶点,作者定制了siRNA文库在ZM-bl细胞系中进行高通量筛选,该文库靶向细胞核内的几种细胞途径的182个基因,包括chromatin binding, epigenetic modification, chromatin remodeling, ubiquitination, SUMOylation 和 chromosome organization。分析敲除各靶标基因后荧光素酶的表达水平变化(以siNC为对照),许多蛋白质对应的荧光素酶表达升高,表明这些蛋白限制HIV-1启动子的活性,最明显包括HP1α、GLP、SUZ12和CYLD,其已被鉴定为抑制HIV-1转录。其中,两个较少见的SUMO化途径基因TRIM28和SUMO4敲低后,显著上调了HIV-1启动子活性。
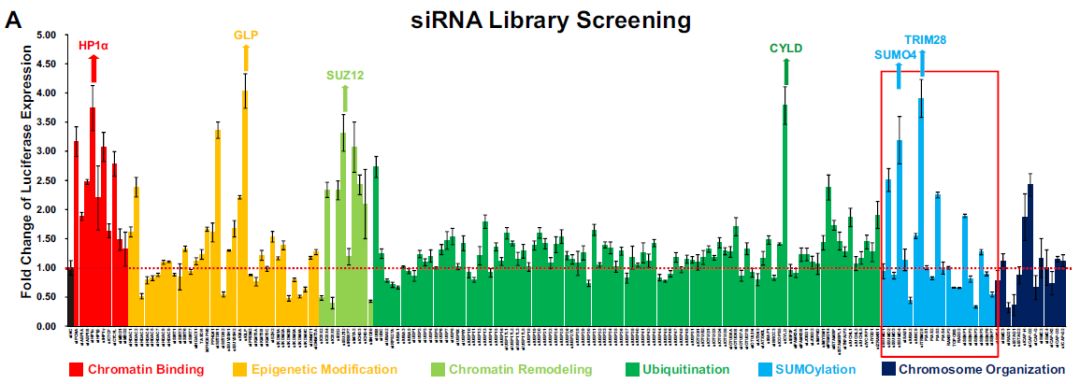
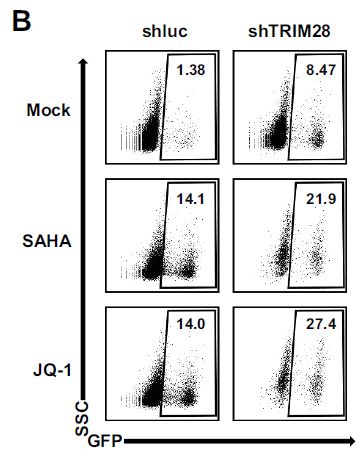
通过实验发现si-TRIM28过表达抑制了HIV-1启动子活性的基础水平,并以剂量依赖的方式挽救了HIV-1抑制;当与HIV-1 Tat和TNFα组合过表达时,上调更显著;TRIM28在多种细胞系和原代细胞中普遍表达。为搜索HIV-1潜伏相关基因,作者利用RNA-Seq比较未刺激和PHA刺激的原代CD4+ T细胞中的基因表达作为补充实验,TRIM28在未刺激的原代CD4+ T细胞中高表达,并在PHA激活后下调。当活化的CD4+ T细胞恢复到静止状态时,TRIM28的表达再次上调。为了测试TRIM28是否有助于HIV-1潜伏,在HIV-1潜伏细胞系J-Lat 10.6以及其他HIV潜伏细胞系模型(J-Lat 6.3/8.4/9.2和15.4)中敲低了TRIM28,发现TRIM28的缺失上调了HIV-1的表达。此外,当补充广泛定义的LRA:组蛋白去乙酰化酶(HDAC)抑制剂SAHA或BET结构域抑制剂JQ-1时,HIV-1重新激活增强得更高。
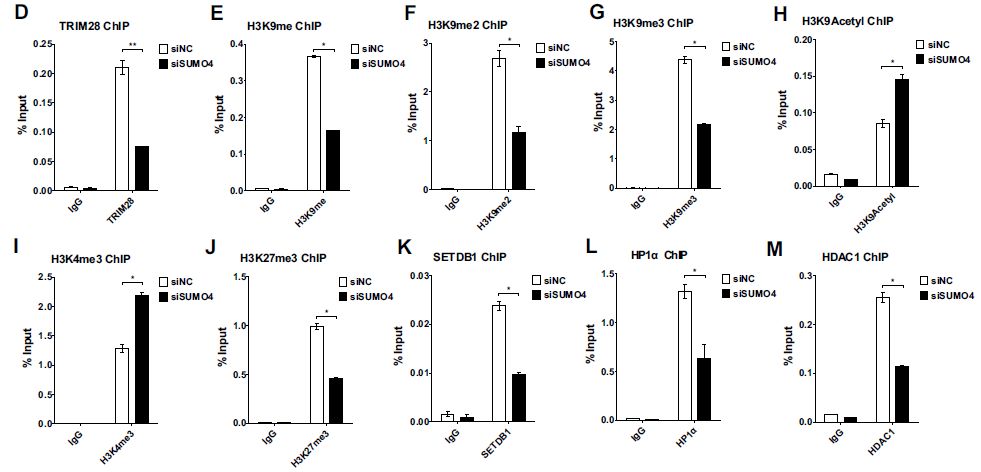
先前已鉴定TRIM28通过募集HDAC1以使HIV-1整合酶去乙酰化来抑制HIV-1整合。在TZM-bl和J-Lat 10.6细胞系中进行TRIM28的染色质免疫沉淀(ChIP)实验,以研究其与整合的HIV-1 DNA的可能关联。结果表明:TRIM28在HIV-1 LTR上显著富集,不受TNFα信号传导的影响。接下来,探究了TRIM28缺失是否会影响HIV-1 LTR的表观遗传状态,敲除TRIM28后H3K9me2和H3K9me3显著减少,以及H3K4me3和H3K9Ac显著增加,也诱导H3K27me3轻微下调,表明TRIM28通过控制抑制性表观遗传修饰来抑制HIV-1表达并导致HIV-1潜伏期。
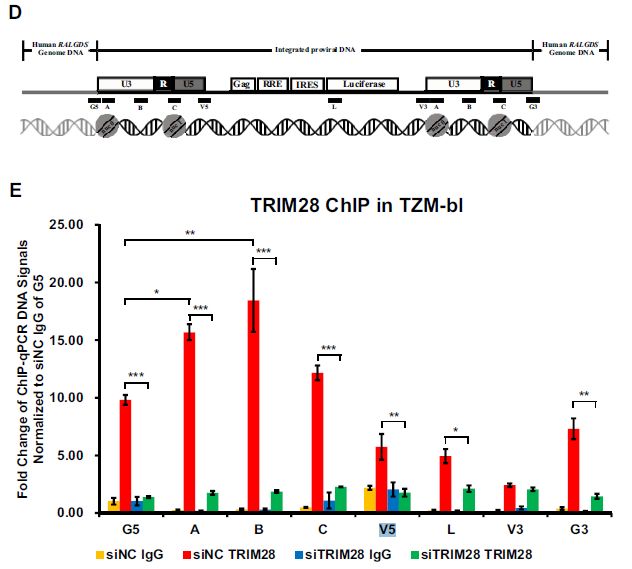
2TRIM28 SUMO化许多转录因子和转移酶
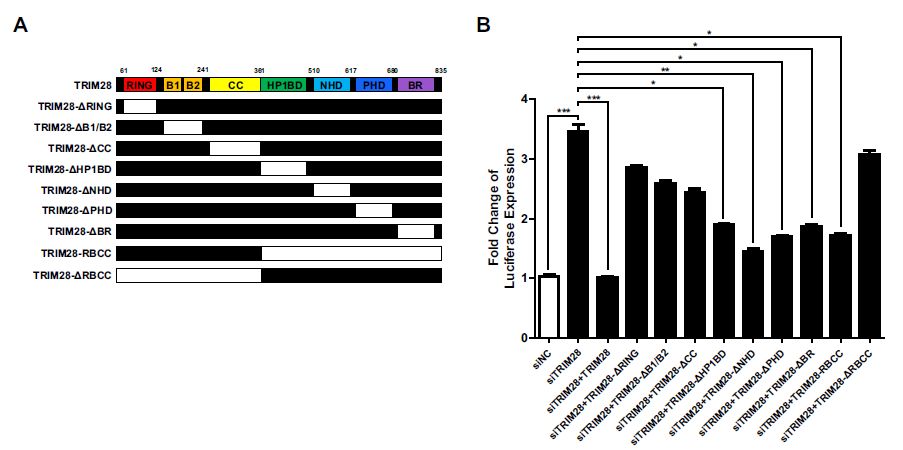
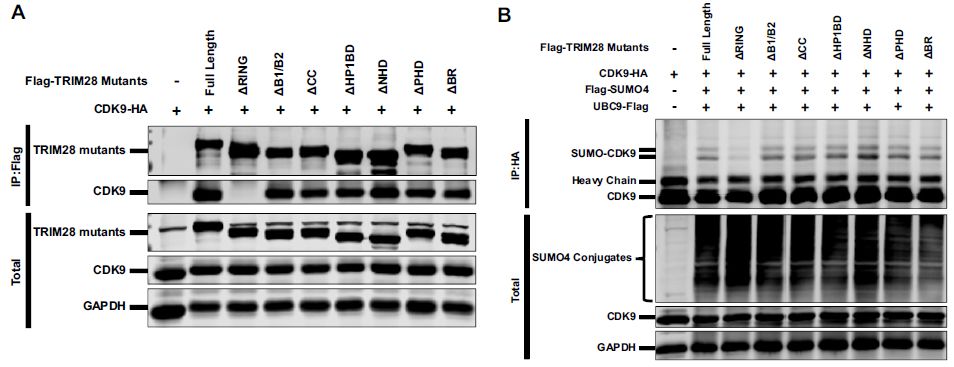
接下来,作者通过构建TRIM28不同区域的缺失突变体探究该蛋白功能。TRIM28是一种多功能蛋白,含有7个不同的结构域:由邻近的植物同源域(PHD)SUMO化的C-末端溴结构域(BR),以SUMOylation依赖性方式募集SETDB1和NuRD复合物;N端三联体基序RBCC区域由RING指结构域(RING)、2个B-box domians(BB)和卷曲螺旋域(CC)组成。 TRIM28的RING起分子间SUMO E3连接酶的作用,而PHD对分子内SUMO E3连接酶活性很重要。
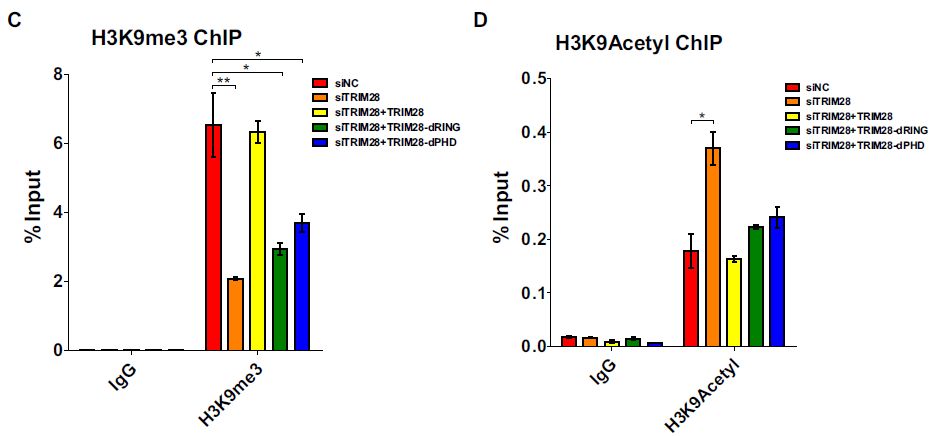
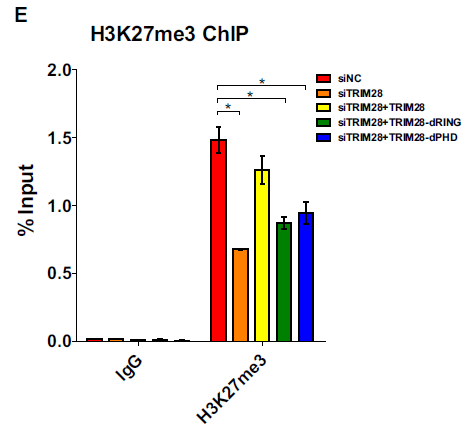
HP1BD、NHD或BR缺失突变体,尤其是具有分子内SUMO E3连接酶活性的PHD突变体,没有能够显著地挽救抑制作用;RING或RBCC结构域缺失突变体完全中止了再抑制,这可能是由于Krüppel相关的盒结构域锌指(KRAB-ZNFs)结合能力的丧失;而仅含有RBCC的突变体(TRIM28-RBCC)仍然恢复了抑制作用。野生型TRIM28能够拯救抑制性表观遗传标记H3K9me3和H3K27me3并抑制活性表观遗传标记H3K9乙酰化,然而,没有RING或PHD结构域的突变体仅能够挽救部分抑制标记。因此,作者假设TRIM28可利用RING结构域SUMO化对HIV-1表达至关重要的细胞蛋白。
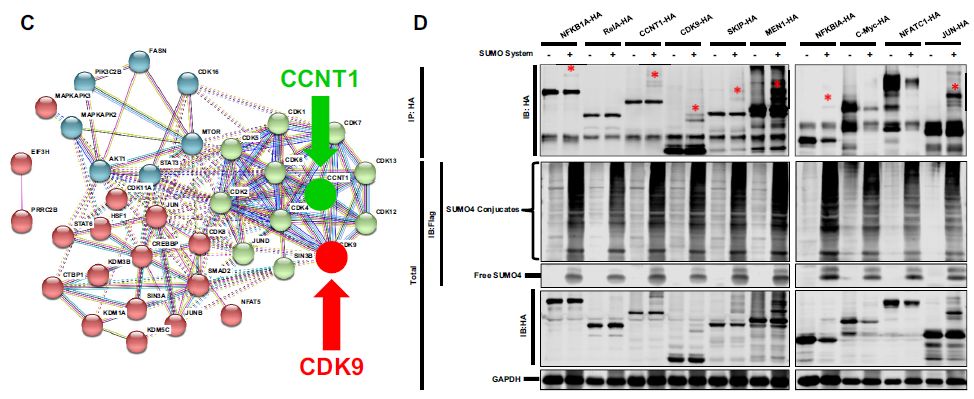
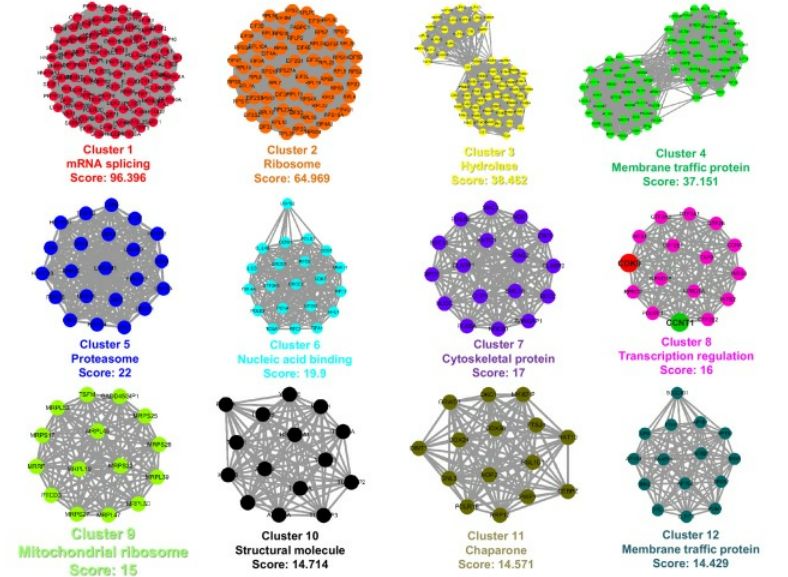
为了鉴定由TRIM28 SUMO化的候选底物,作者进行了全局位点特异性SUMO化质谱(SUMO-MS),鉴定了1,329个SUMO化蛋白,并构建了相互作用置信度为0.7的STRING互作网络,MCODE分析发现STRING核心网络可以聚类成12个亚群。GO分析发现细胞和代谢过程是SUMO化蛋白可能参与的前两个生物过程。通过k-means聚类特异性地将转移酶和转录因子聚类,并用STRING分析进行可视化,发现许多候选蛋白对于HIV-1表达是关键的,例如JUN、JUNB、JUND、mTOR、STAT3、Cyclin T1(CCNT1)和CDK9。将SUMO化蛋白鉴定的显著性阈值调低到10^-8,CDK9仍然排名靠前。作者在体外验证了全局位点特异性SUMO-MS的可靠性。
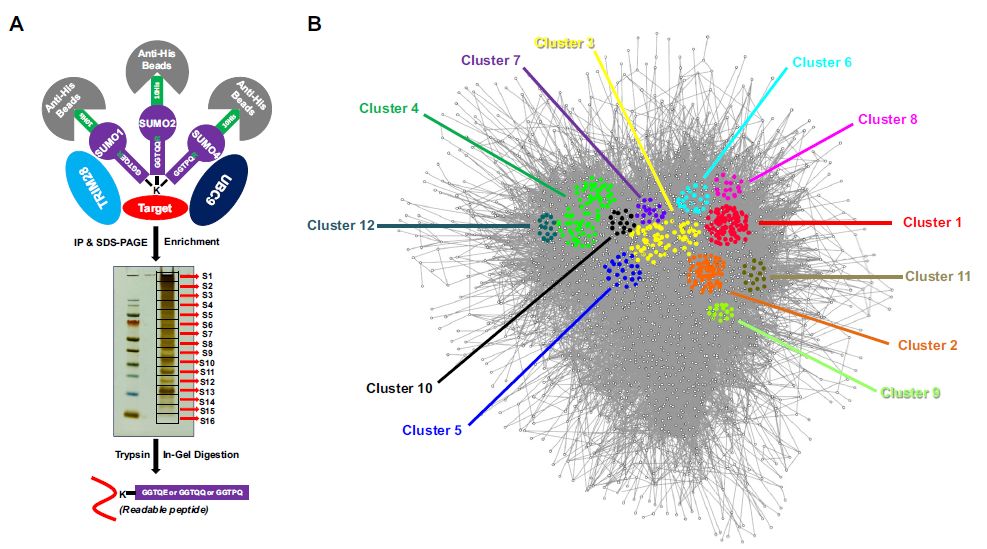
3、CDK9由TRIM28 SUMO化
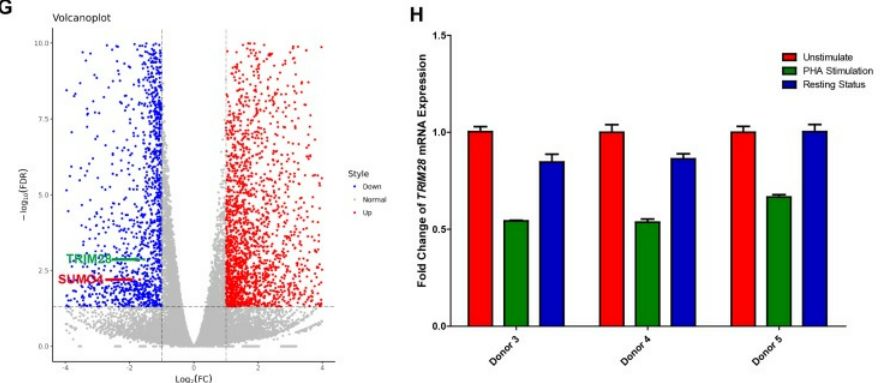
前面siRNA文库筛选中,SUMO4缺失比其他SUMO旁系同源物缺失更显著上调HIV-1启动子活性。当与HIV-1 Tat过表达组合时,上调更显著,其现象与TRIM28实验中观察到的相似。SUMO4的敲低或敲除也能够在J-Lat 10.6中重新激活潜在的假HIV-1。原代CD4+ T细胞被PHA刺激后,SUMO4的表达下调,恢复到静止期后达SUMO4表达恢复到基础水平。
接下来探究了SUMO4是否可以影响TRIM28的功能和HIV-1启动子的表观遗传状态:SUMO4敲低后,超过一半的TRIM28从HIV-1 LTR中丢失,表明TRIM28在HIV-1 LTR上的富集可能是部分SUMOylation依赖性的;SUMO4敲低后H3K9me、H3K9me2和H3K9me3在HIV-1 LTR上显著降低,以及H3K9甲基化’writer’ SETDB1和’reader’ HP1α显著降低;H3K9乙酰化和H3K4me3的显著上调和HDAC1的下调,这与先前报道TRIM28以SUMO化依赖性方式募集SETDB1、HP1α和HDAC1一致;H3K27me3在HIV-1 LTR上也降低。一些多梳抑制复合物2(PRC2)组分如EZH2和SUZ12(H3K27me3的主要’writer’)可能被SUMO4 SUMO化,导致修饰功能的增强。
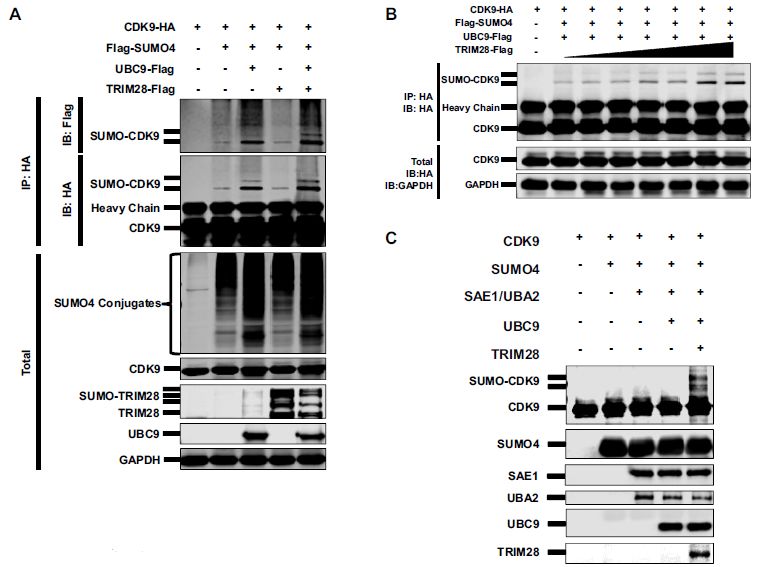
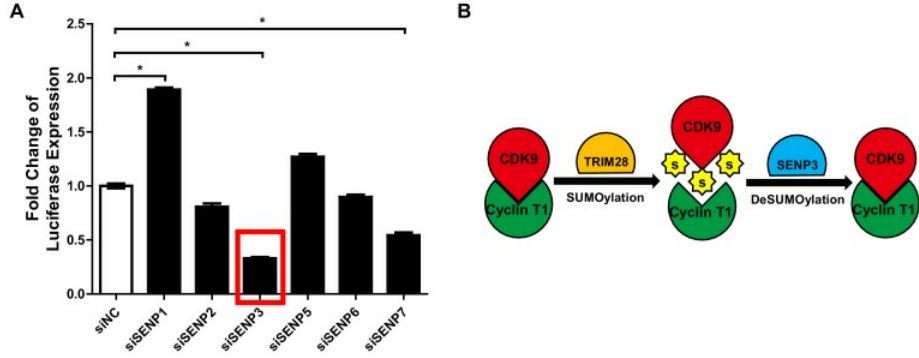
作者进一步研究SUMO4在TRIM28介导的CDK9 SUMO 经中的作用来鉴定其潜在的机制。通过CDK9与SUMO1、SUMO2和SUMO4共同过表达实验,发现CDK9主要由SUMO1和SUMO4 SUMO化。补充过表达TRIM28后,SUMO-CDK9量变得更丰富。然而,如果仅用TRIM28共表达CDK9但没有SUMO E2 酶UBC9时,SUMO化水平没有增加,这表明TRIM28介导的SUMO化是UBC9依赖性的。当TRIM28表达逐渐增加时,SUMO-CDK9 化水平以剂量依赖性增加。体外SUMO化测定表明,仅当将SUMO4、E1 SAE1/UBA2,E2 UBC9和TRIM28一起提供到SUMO化反应缓冲液中时,SUMO4与CDK9结合。在HeLa细胞中敲除TRIM28后,SUMO化的CDK9减少。前面的siRNA文库筛选实验中表明,几种SUMO特异性蛋白酶(SENP)抑制HIV-1表达,尤其是SENP3,用TRIM28共表达SENP3,发现SENP3阻止了TRIM28介导的CDK9 SUMO化。作者在原代CD4+ T细胞中证实了以上结论。总之,TRIM28介导SUMO4与CDK9的结合,其被SENP3逆转。
4、TRIM28 RING结构域在结合和SUMO化CDK9中起关键作用
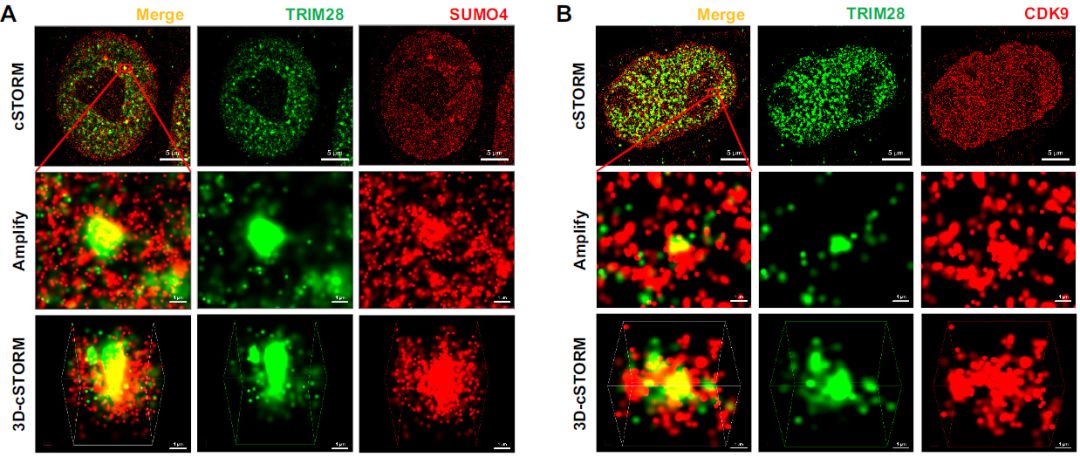
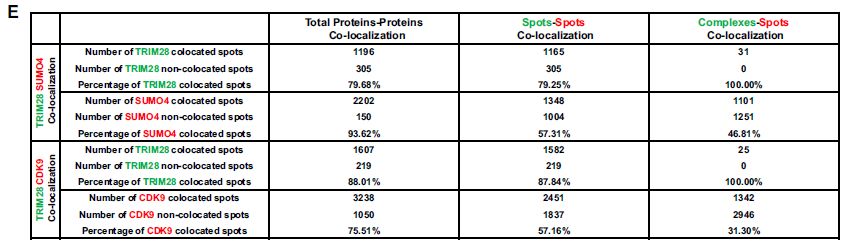
为了确定TRIM28是否与CDK9结合,作者使用超分辨率的连续随机光学重建显微镜(cSTORM)来研究分辨率为20nm二者间的三维(3D)共定位。TRIM28存在于细胞核中,与点状SUMO4共定位。放大视图和3D-cSTORM,发现SUMO4蛋白被TRIM28富集并形成大斑点。CDK9存在于细胞核内的分散点中,但仍发现CDK9与TRIM28共定位。与SUMO4类似,CDK9蛋白被TRIM28富集并包围TRIM28。近80%的TRIM28斑点或复合物与94%的SUMO4斑点共定位,88%的TRIM28斑点或复合物与76%的CDK9斑点共定位。
免疫共沉淀(Co-IP)实验发现即使在RNase存在下CDK9也与TRIM28结合。为了鉴定TRIM28的哪个区域与CDK9结合,构建各种TRIM28缺失突变体以富集CDK9:RING结构域缺失中止了CDK9的结合以及CDK9的SUMO化。实验表明外源表达的TRIM28也与CDK9共定位。总之,研究表明RING和PHD都具有E3连接酶活性并富集UBC9。

5、TRIM28 SUMO化时抑制CDK9功能
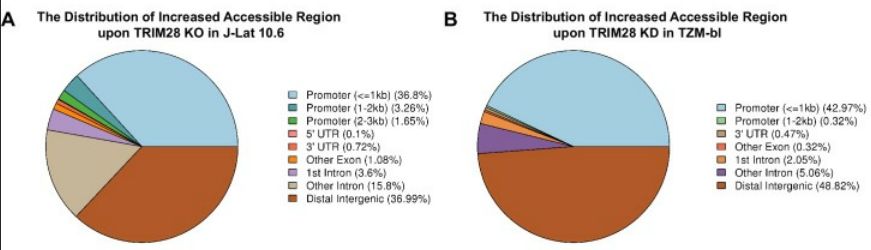
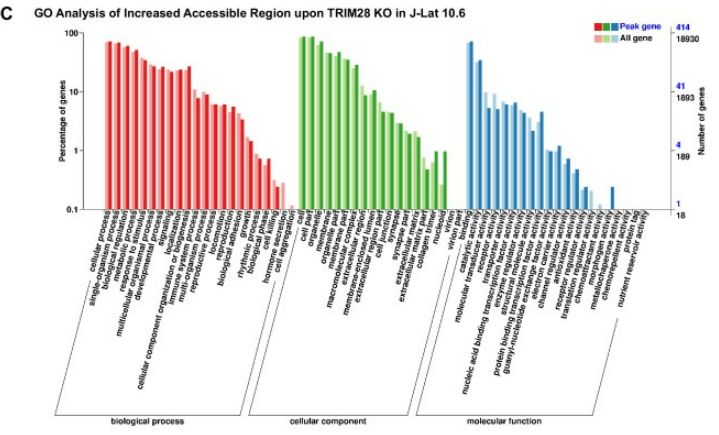
进一步探究CDK9的功能是否受TRIM28介导的SUMO化的影响。首先,ATAC-Seq探究TRIM28缺失后HIV-1启动子的染色质可及性。在J-Lat 10.6或TZM-bl细胞系中TRIM28缺失时,基因组中大多数增加的可及区域位于启动子和远端基因间区域上。GO分析和COG分析表明,染色质可及性变化发生在与各种生物过程和细胞组分相关的基因中,大多数受影响的功能基因具有DNA或蛋白质结合能力和催化活性。
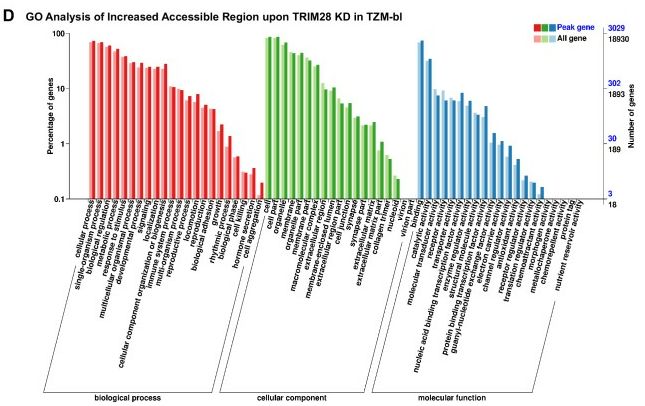
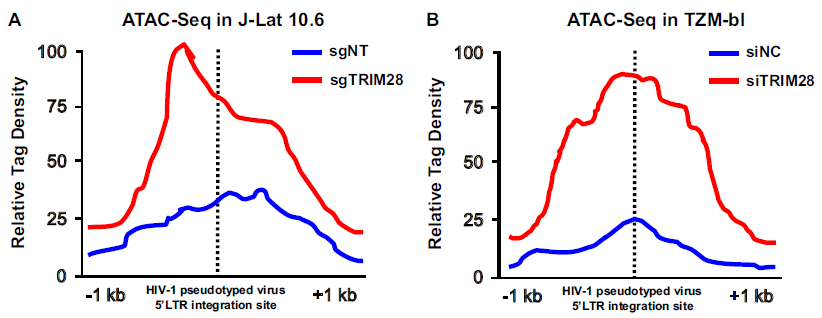
作者也对HIV-1基因组的染色质可及性是否受到TRIM28缺失的影响进行了分析,将ATAC-seq数据以HIV-1参考基因组进行分析,转座标签密度所指示的可及区域在HIV-1 LTR上增加,这表明启动子活性显著增强。且敲除TRIM28或SUMO4后HIV-1 LTR上CDK9和Ser2超磷酸化RNAP II显著富集,这与TRIM28或SUMO4缺失重新激活HIV-1表达的结果一致。
Co-IP实验发现细胞周期蛋白Cyclin T1仅与野生型CDK9结合形成正性转录延伸因子P-TEFb复合物,而不是SUMO化的CDK9。为了研究TRIM28介导的CDK9 SUMO化是否影响CDK9的激酶活性,进行了体外CDK9 SUMO化测定和CDK9激酶活性测定:发现当TRIM28 SUMO化CDK9时,CDK9的激酶活性显著降低。不添加TRIM28,CDK9的激酶活性不受影响,尽管已添加其他SUMO化组分。
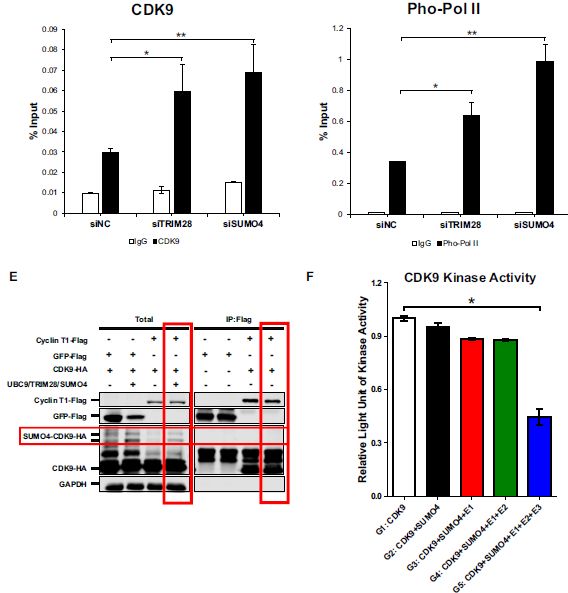
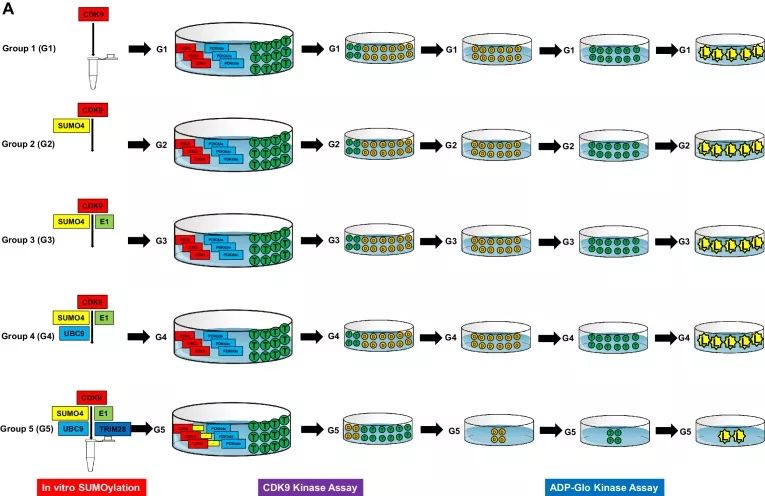
总之,TRIM28介导的SUMO化损害了CDK9与Cyclin T1的结合能力和CDK9对RNAP II的激酶活性,导致转录延伸功能障碍。
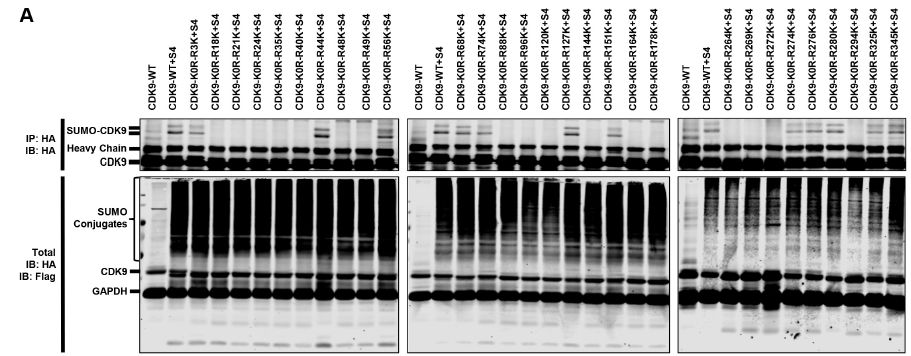
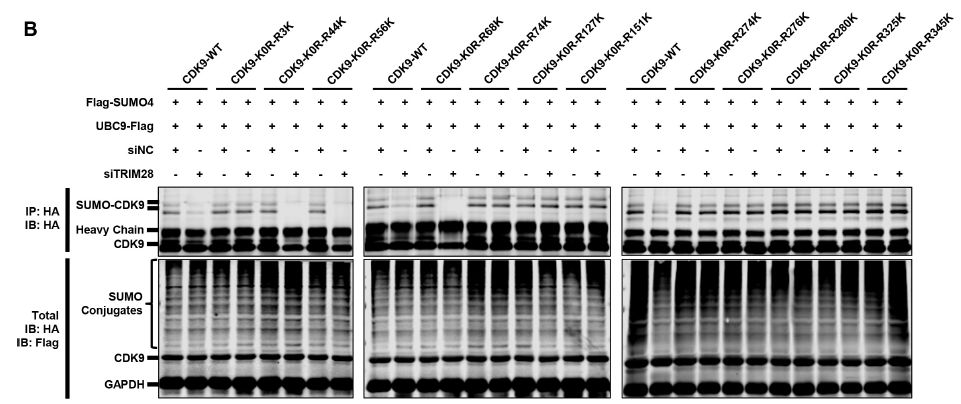
6、SUMO4对CDK9的Lys44\Lys56\Lys68残基SUMO化
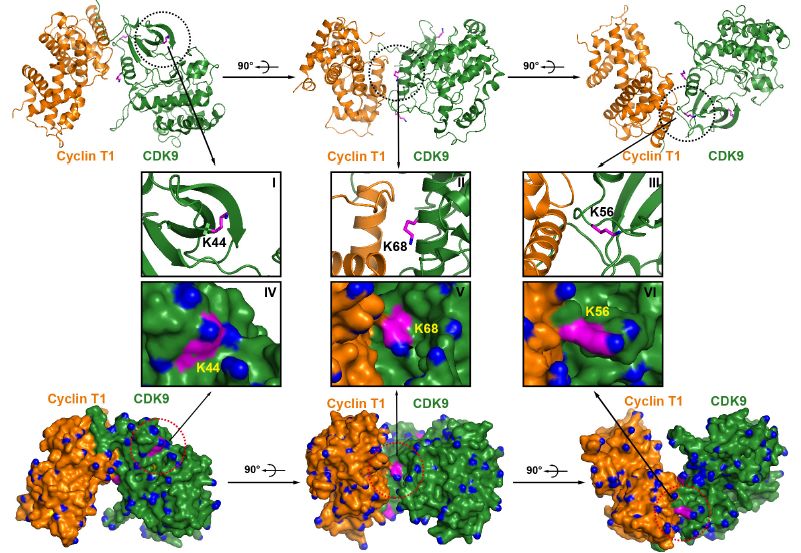
接下来作者试图鉴定应该在CDK9 赖氨酸残基上发生的SUMO化位点。赖氨酸突变实验结果:CDK9-K0R突变体含有所有赖氨酸变为精氨酸的突变,完全中止了CDK9被SUMO化的能力;其他CDK9突变体仍然能够通过TRIM28进行SUMO化,这表明在整个CDK9序列中可能存在多个SUMOylation位点。采用基于CDK9-K0R全突变体的逆转突变策略,发现CDK9上的几个赖氨酸显著SUMO化。其中,多个SUMO化位点与CDK9 C-末端自身磷酸化位点相邻,据报道这些位点是Tat-P-TEFb与TAR RNA高亲和力结合所必需的,即 SUMO化可以通过阻止相邻的磷酸化来降低结合能力。
为了进一步确定哪些位点确实仅被TRIM28 SUMO化,作者敲除了内源性TRIM28并测试了上述鉴定候选位点的SUMO化潜力,发现Lys44、Lys56和Lys68的SUMO 化信号在缺乏内源性TRIM28时完全消失,进一步支持这些位点被TRIM28特异性SUMO化。直接分析富集的SUMO-CDK9的靶特异性SUMO-MS也证实了该结果。Lys44的乙酰化是其激酶活性所必需的,其他2个SUMO化位点Lys56和Lys68在CDK9和细胞周期蛋白T1的相互作用区域内。SUMO蛋白是多肽大分子,它的存在可以形成空间位阻,从而阻止P-TEFb复合物的形成。

7、TRIM28缺失重新激活HIV-1感染者细胞中潜伏HIV-1
为了验证TRIM28是否可以成为开发新LRA的安全靶标,首先对Hela细胞、Jurkat细胞以及从感染者分离的静息CD4+ T细胞中评估了缺失TRIM28相关的可能毒性:细胞毒性测定、细胞活性测定、细胞数计数和细胞增殖测定。结果表明TRIM28缺失对细胞活性和增殖无害。
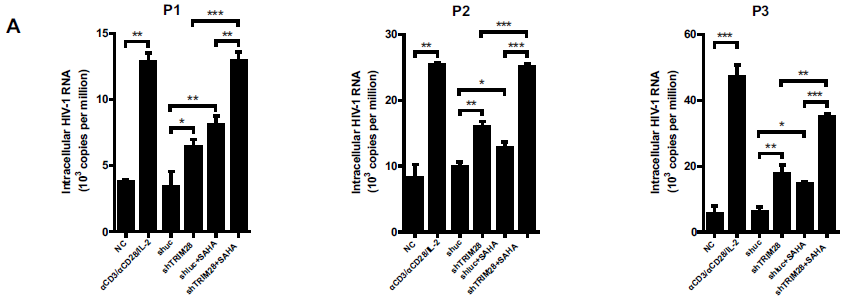
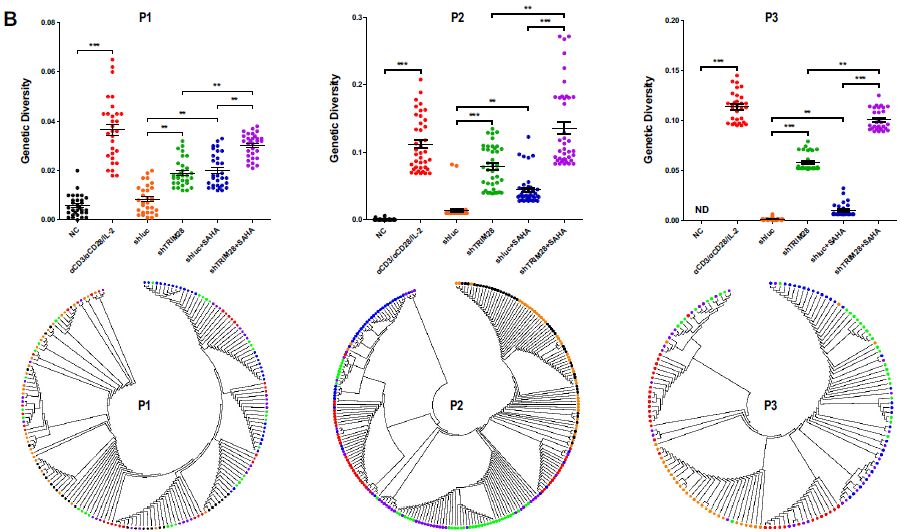
之后作者试图确定TRIM28的敲低是否在接受抑制性cART疗法至少6个月的HIV-1感染者的静息CD4+T细胞中重新激活潜伏的HIV-1。基于细胞内HIV-1 RNA的定量,表明用αCD3/αCD28刺激显著诱导HIV-1的表达。TRIM28敲低重新激活了与HDAC抑制剂SAHA相似量的HIV-1 RNA表达。在将TRIM28敲低与SAHA组合后,重新激活效果更显著。同样,SUMO4敲低和SAHA添加的组合使用可以重新激活比单独使用更多的HIV-1 RNA表达。虽然TRIM28单独缺失重新激活相似数量的具有SAHA的遗传多样化HIV-1表达,但TRIM28敲低和SAHA的组合重新激活了更多遗传多样化的HIV-1表达。
为了确定重新激活的HIV-1是否具有复制能力,用PHA激活的健康个体分离的CD4+ T共培养了HIV-1感染个体的PHA刺激的、SAHA诱导的或TRIM28敲低的静息CD4+ T细胞。p24抗原的累积产生表明重新活化的HIV-1病毒颗粒具有复制能力。TRIM28的敲低在所有3个样品中重新激活了具有复制能力的病毒。类似地,SAHA与TRIM28敲低的组合重新激活了更多具有复制能力的病毒颗粒。这些结果表明TRIM28有助于HIV-1感染个体的HIV-1潜伏,靶向TRIM28对HIV-1感染的CD4+ T细胞具有良好的适用性。
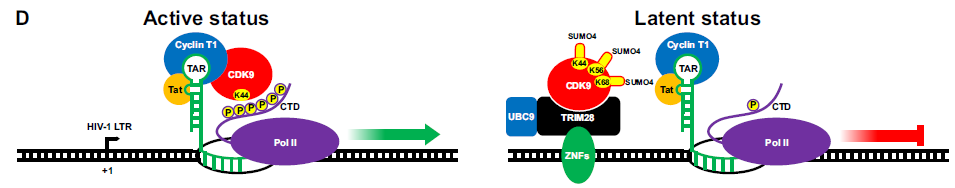
作者发现TRIM28不仅可以作为定义明确的表观遗传学adaptor,还可以作为SUMO E3连接酶与SUMOylate P-TEFb复合物一起发挥作用,从而显著抑制HIV-1的表达并导致HIV-1潜伏。提出了TRIM28介导的HIV-1潜伏模型:在活跃状态下,P-TEFb复合物被HIV-1 Tat募集至部分转录的HIV-1 RNA反式激活反应元件(TAR)。 P-TEFb催化亚基CDK9使RNAP II的Ser2残基超磷酸化,促进RNAP II转录HIV-1 RNA的持续性;在潜伏状态下,TRIM28被募集到HIV-1 LTR并在Lys44、Lys56和Lys68 SUMO化CDK9,导致CDK9激酶活性的抑制和其与细胞周期蛋白T1的隔开,没有Ser2的超磷酸化,RNAP II启动子近端暂停在LTR。因此,潜伏状态由TRIM28介导的CDK9功能障碍和TRIM28介导的核小体nuc-1的抑制性表观遗传修饰维持,核小体nuc-1恰好位于HIV-1启动子的下游。
参考文献:
Ma X, Yang T, Luo Y, et al. TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb.Elife. 2019 Jan 17;8. pii: e42426.
如果您的项目有任何问题,欢迎点击下方按钮联系我们。





 京公网安备 11011302003368号
京公网安备 11011302003368号