


研究背景
表皮生长因子受体酪氨酸激酶抑制剂 (EGFR-TKI) 是癌症靶向治疗的经典例子。已获批的 EGFR-TKIs 单独使用或与化疗联合用于治疗非小细胞肺癌 (NSCLC)、结直肠癌 (CRC)、乳腺癌 (BC) 等。然而,大多数患者在 1-2 年后对 EGFR-TKI 产生获得性耐药,随后癌症复发和转移。为了解决这一紧迫的临床问题,迫切需要延迟和克服 EGFR-TKIs 耐药的策略。到目前为止,获得性 EGFR 突变、MET 扩增、ERBB2 扩增和旁路激活等作为 EGFR-TKI 耐药的机制已得到充分研究。近年来,一些研究人员开始关注肿瘤免疫环境状态与 EGFR-TKIs 治疗疗效之间的关系。临床研究最近发现,EGFR-TKIs 治疗可以重塑肿瘤免疫环境,但 EGFR-TKIs 与瘤周免疫细胞之间的关系尚未得到充分研究。
实验材料
使用携带 EGFR 突变的小鼠细胞系建立了异位异种移植肿瘤模型,并评估了对不同代 EGFR-TKI 的反应,揭示肿瘤耐药机制。
进行天然小分子化合物库 (MCE) 的筛选,制作了 266 个样品。筛选 PD-1/PD-L2 结合抑制剂。
研究结果
-
肿瘤免疫环境参与肿瘤对 EGFR-TKIs 的耐药性
首先,该研究使用携带 EGFR 突变的小鼠细胞系建立了异位异种移植肿瘤模型,并评估了对不同代 EGFR-TKI 的反应。EGFR 突变或扩增存在于多种癌症中,如非小细胞肺癌、结直肠癌、乳腺癌等。EGFR-TKIs 可以靶向 EGFR 敏感突变。因此,作者使用小鼠肺腺癌细胞 (LLC) 和结直肠癌细胞 (CT26) 作为工具细胞。作者用突变形式的人 EGFR 转染小鼠细胞系 (LLC 和 CT26),携带外显子 19 缺失 (EGFR19del) 或外显子 21 L858R 错义突变 (EGFRL858R),这会增加 EGFR 活性,以接近细胞模型到人类疾病,建立同基因荷瘤小鼠模型。在转染 EGFR 突变体的细胞中,p-EGFR 和 p-ERK1/2 的表达增强,增殖速率显著增加,表明细胞 EGFR 下游信号通路激活(图 1A、B)。正如预期的那样,EGFR 突变细胞对不同代 EGFR-TKI 更敏感,包括厄洛替尼、阿法替尼和奥希替尼(图 1B)。为了全面探讨 EGFR-TKIs 耐药的机制,作者使用 EGFR 突变细胞建立了第一代和第三代 EGFR-TKIs 耐药同基因小鼠模型(图1C)。如图1 D所示,第一代 (P1) LLC-EGFR19del 来源的肿瘤对奥希替尼敏感 (抑制率 = 44.0%)。在第 2 代 (P2) 中,作者将奥希替尼的剂量增加了一倍,肿瘤抑制率降低了一半 (22.2%)。用奥希替尼治疗的第三代 (P3) LLC-EGFR19del衍生肿瘤被认为对奥希替尼耐药,因为它们的体积与用生理盐水治疗的小鼠的体积没有显着差异。同时,作者还使用类似的方法成功获得了对奥希替尼耐药的 LLC-EGFRL858R衍生肿瘤和对厄洛替尼耐药的 CT26-EGFR19del衍生肿瘤(图 1D)。
为了进一步研究 EGFR-TKIs 耐药与肿瘤免疫微环境之间的关系,作者将 P3 CT26-EGFR19del衍生的肿瘤接种到免疫功能正常的 BALB/c 小鼠和严重免疫缺陷的 NOD (PRKDCKO IL2RKO) 小鼠中,然后作者评估了它们对厄洛替尼的反应性(图 1E)。作者的结果表明,接种到 BALB/c 小鼠中的肿瘤对厄洛替尼治疗保持耐药性(抑制率 = 6.9%),而接种到 NOD (PRKDCKO IL2RKO) 小鼠中的肿瘤显示出降低的耐药性(抑制率 = 40.2%)(图1E)。与体内肿瘤生长数据一致,增殖生物标志物 Ki67 的表达在用厄洛替尼处理后也显示免疫缺陷异种移植肿瘤组织显着减少,但在免疫活性异种移植肿瘤组织中没有减少(图 1F)。总之,这些结果表明肿瘤免疫环境参与 EGFR 突变肿瘤对 EGFR-TKI 的耐药性。
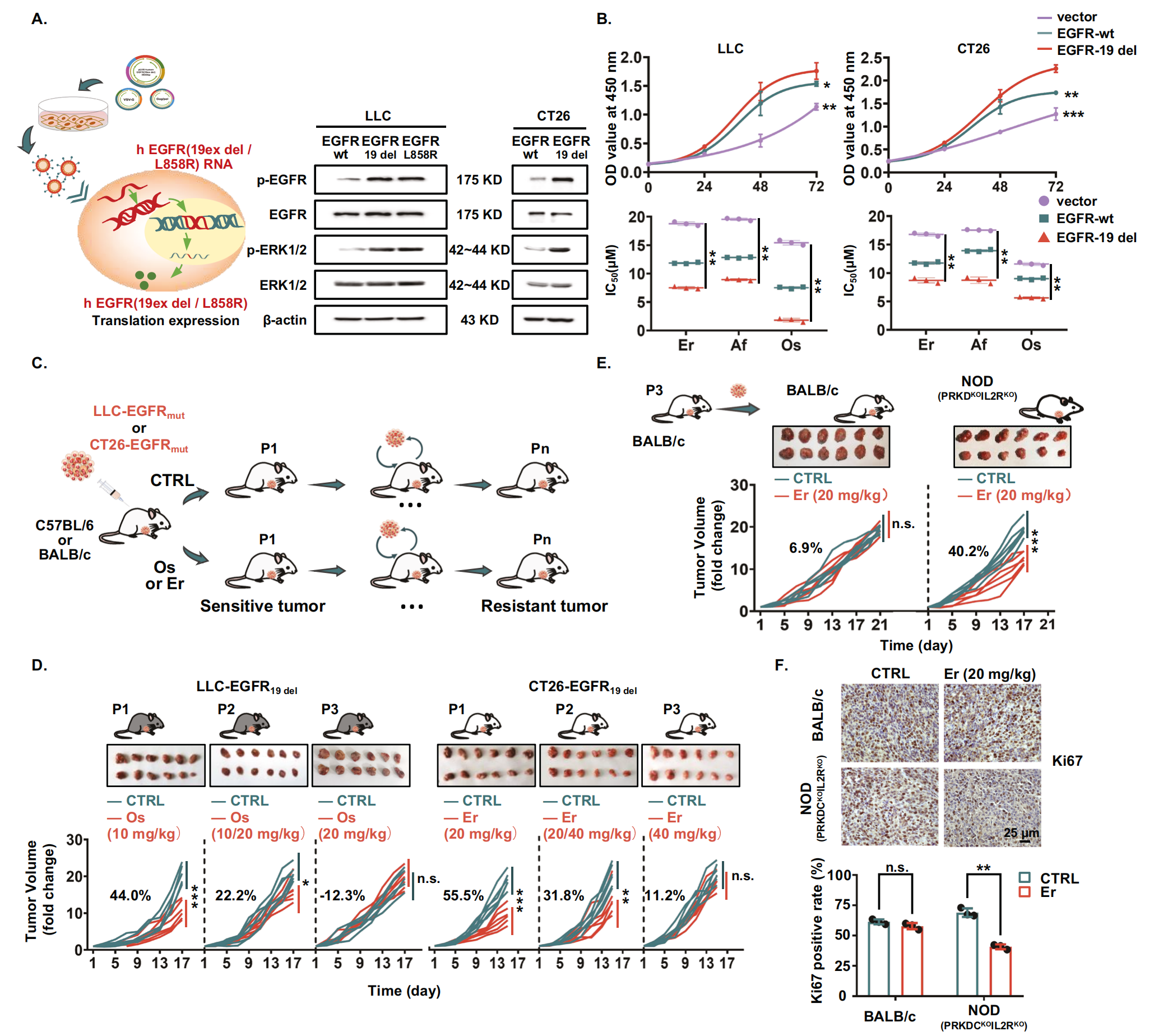
图1-肿瘤免疫环境参与肿瘤对 EGFR-TKIs 的耐药性
- 肿瘤免疫环境由激活到抑制的动态变化驱动 EGFR-TKIs 耐药
为了了解肿瘤免疫环境如何参与对 EGFR-TKI 的耐药性,作者分析了治疗过程中不同阶段的肿瘤免疫环境特征(图 2A)。作者测定了初始 EGFR-TKIs 治疗时和耐药发展期间 (P1-P3) 肿瘤免疫环境中肿瘤浸润白细胞 (TIL) 的密度,其中用盐水处理的肿瘤细胞作为 CTRL 组,用厄洛替尼或奥希替尼治疗的肿瘤细胞作为 TKI 组。流式细胞术分析的免疫分析显示,在治疗早期,携带 EGFR19del 的肿瘤的 EGFR-TKIs 治疗诱导肿瘤浸润白细胞比例增加(图2B)。然而,当荷瘤小鼠对 EGFR-TKI 产生耐药性时,与 CTRL 组相比,肿瘤浸润白细胞的比例显著降低(图2B)。这表明荷瘤小鼠对 EGFR-TKIs 的反应性改变伴随着肿瘤免疫环境的动态变化。
为了准确反映肿瘤免疫环境的真实状态,作者检查了药物治疗期间荷瘤小鼠肿瘤免疫环境中的 T 细胞浸润和功能。作者观察到,两种模型的 P1 荷瘤小鼠中 CD8+ T 细胞比例均显著增加,但随着 TKI 治疗的继续,这种增加消失。TKI 耐药 P3 荷瘤小鼠中 CD8+ T 细胞的比例显著降低(图2C)。因此,作者假设 T 淋巴细胞,尤其是 CD8+ T 细胞,从最初的激活开始逐渐获得功能失调的表型。为了检验这一假设,作者检查了效应细胞因子在 T 细胞 (IFN-γ、IL-2、颗粒酶 B 和 TNF-α)中的表达。EGFR-TKIs 的初始治疗刺激效应细胞因子的产生,但在耐药发展的后期,效应细胞因子的水平似乎降低,并且与 CTRL 相比,IFN-γ 和颗粒酶 B 的产生显着减少(图2D, E)。这些结果表明 EGFR-TKIs 显着影响肿瘤免疫环境。初始治疗会引发有效的抗肿瘤免疫反应,从而显着减少肿瘤生长。随着时间的推移,EGFR-TKI 无法显著控制肿瘤生长,并伴有 T 细胞浸润和细胞因子产生的显著减少。综上所述,这些结果表明,随着 EGFR-TKIs 治疗时间的延长,肿瘤细胞由药物敏感变为耐药,免疫反应首先增强后抑制。
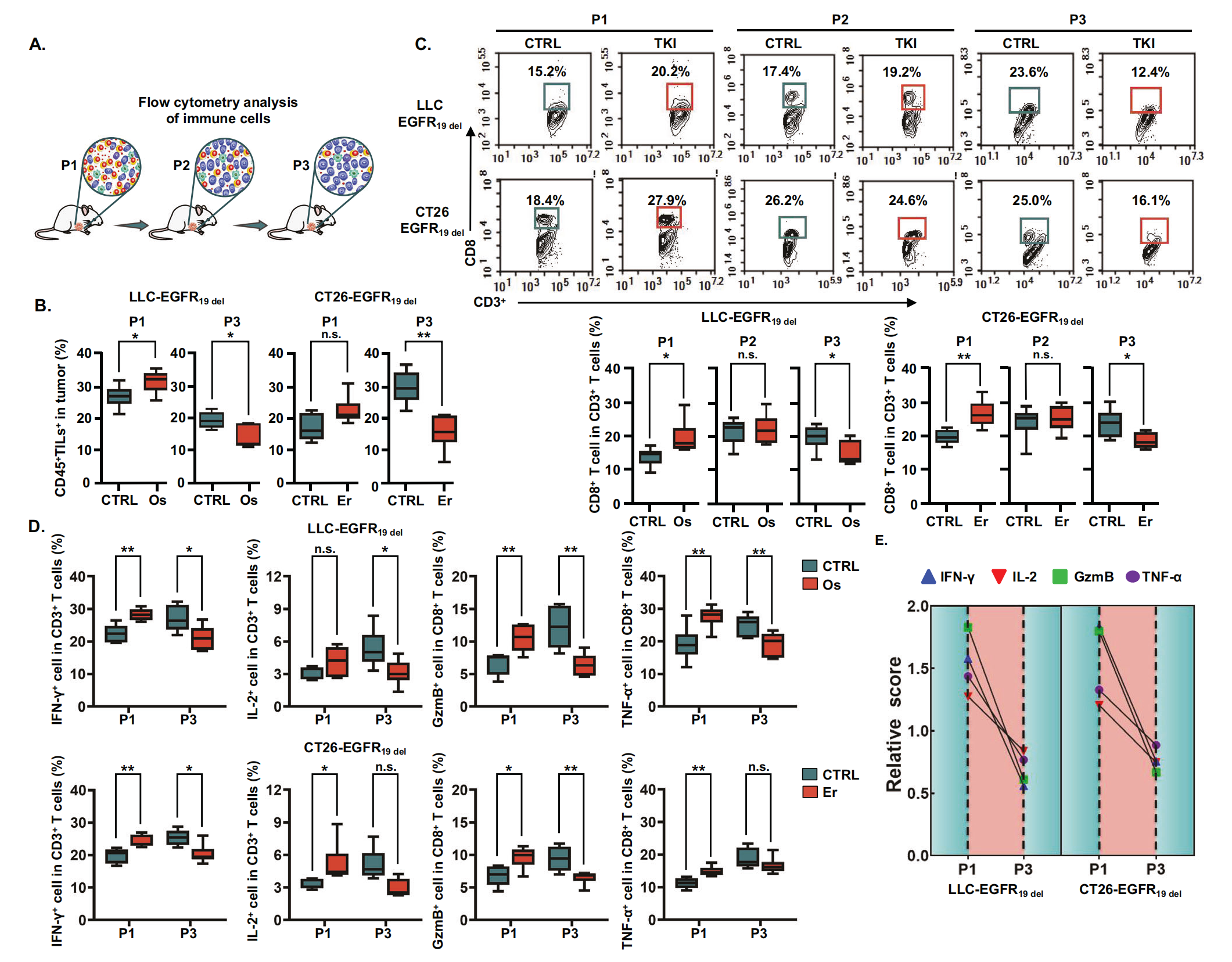
图2-肿瘤免疫环境由激活到抑制的动态变化驱动 EGFR-TKIs 耐药
- 对 EGFR-TKI 的获得性耐药伴随着 PD-L2 表达的上调
为了探索肿瘤免疫环境变化的原因,作者同时检测了不同传代阶段肿瘤组织中刺激性和抑制性免疫调节分子的表达。在 EGFR-TKI 的初始治疗中,一些抑制性免疫检查点的相对表达降低(图随着耐药性的出现,抑制性免疫检查点基因 PD-L1 和 PD-L2 的相对表达水平逐渐升高,尤其是 PD-L2 (图3A)。为了进一步验证 PD-L2 在 EGFR-TKI 耐药肿瘤细胞中表达的增加,作者通过流式细胞术鉴定了 PD-L2 在 P3 肿瘤细胞厄洛替尼或奥希替尼处理的肿瘤细胞(耐药,TKI 组)表面的表达,以生理盐水处理的细胞为对照(敏感,CTRL 组)。作者的结果表明,与对照组织相比,肿瘤细胞表面 PD-L2 的表达在耐药肿瘤组织中表现出显着增加(图3B)。接下来,为了进一步验证,作者分析了先前构建的 EGFR 突变的 TKI 耐药 NSCLC 细胞系 HCC4006R、HCC827R、PC9R 及其配对亲本细胞中免疫检查点标志物的表达水平。在三种耐药细胞系中,PD-L2 的 mRNA 和蛋白表达水平显著上调,而 PD-L1 的表达表现出不一致的变化(图3C、D)。

图3-EGFR-TKI 的获得性耐药伴随着 PD-L2 表达的上调
为了探索这种现象的临床价值,作者分析了临床数据库。使用 GEO 数据集,作者分析了 8 例 EGFR TKIs 治疗前后切除的临床患者肺癌组织中 PDCD1LG2 (编码 PD-L2) 和其他免疫检查点基因的变化。与作者的结果类似,作者观察到 EGFR TKI 治疗后患者组织中 PDCD1LG2 表达增加,但其他免疫检查点相关基因没有增加(图3E)。此外,作者认为其他因素,例如 MET 扩增,可能涉及 EGFR-TKI 耐药。作者利用 TCGA 数据集研究了 EGFR-TKIs 耐药驱动基因的表达与人 CD274 (编码 PD-L1) 或 PDCD1LG2 表达之间的关系。计算各驱动基因表达水平与 CD274 或 PDCD1LG2 表达水平之间的相关系数。结果表明,PD-L2 表达而不是 PD-L1 表达与 EGFR-TKIs 耐药驱动基因的表达呈显著正相关(图 3F)。与体外实验中的 PD-L1 过表达细胞相比,PD-L2 过表达的 PC9 细胞对上述耐药驱动基因相关的抑制剂的抵抗力增强(图3F)。这些结果与一些研究表明 EGFR-TKIs 耐药机制与 PD-L1 表达有关的研究形成鲜明对比。总而言之,作者的结果提供了初步证据,表明 PD-L2 而不是 PD-L1 可能是介导 EGFR-TKIs 治疗过程中肿瘤环境免疫抑制作用和驱动耐药的关键分子。
- PD-L2 通过影响肿瘤免疫环境在介导 EGFR-TKIs 耐药中发挥关键作用
为了研究 PD-L2 表达在免疫系统中的独特作用和对 EGFR-TKI 的反应,作者在 CT26-EGFR19del 细胞系中过表达 PD-L1 和 PD-L2,并建立了 BALB/c 同基因小鼠模型(图 4A)。使用携带空载体的 CT26-EGFR19del细胞系作为对照。作者通过测量肿瘤体积评估空载体组、PD-L1 过表达组和 PD-L2 过表达组小鼠肿瘤对厄洛替尼治疗的敏感性。结果显示,与空载体组相比,PD-L2 过表达组小鼠对厄洛替尼的肿瘤耐药性显著增加,但 PD-L1 过表达组对厄洛替尼的肿瘤耐药性没有增加(图4B,C)。这些结果表明,PD-L2 的过表达会增加肿瘤细胞对 EGFR-TKIs 的耐药性。为了进一步证实作者的发现,作者检查了 PD-L1 或 PD-L2 过表达对 EGFR-TKIs 治疗荷瘤小鼠肿瘤免疫环境中 T 细胞浸润和细胞因子产生的影响。结果显示,CD4+ T 细胞占总 CD3+ T 细胞的比例在 3 组间差异不显著。值得注意的是,与空载体组相比,PD-L2 过表达组的 CD8+ T 细胞占总 CD3+ T 细胞的百分比以及细胞因子 IFN-γ 和颗粒酶 B 的表达显著降低(图 4D)。然而,PD-L1 过表达组中的细胞因子 IFN-γ 和颗粒酶 B 没有显着变化(图 4D)。免疫组化染色显示,与空载体和 PD-L1 过表达组相比,PD-L2 过表达组肿瘤细胞增殖显著增加(图4E)。以上结果进一步表明,PD-L2 在厄洛替尼治疗期间在抑制免疫反应、抑制细胞毒性 T 细胞浸润和减少小鼠效应细胞因子产生方面起主导作用。因此,作者得出结论,在 EGFR-TKIs 治疗过程中,PD-L2 水平升高通过介导肿瘤细胞的免疫逃逸来抑制肿瘤细胞对 EGFR-TKIs 的治疗反应。
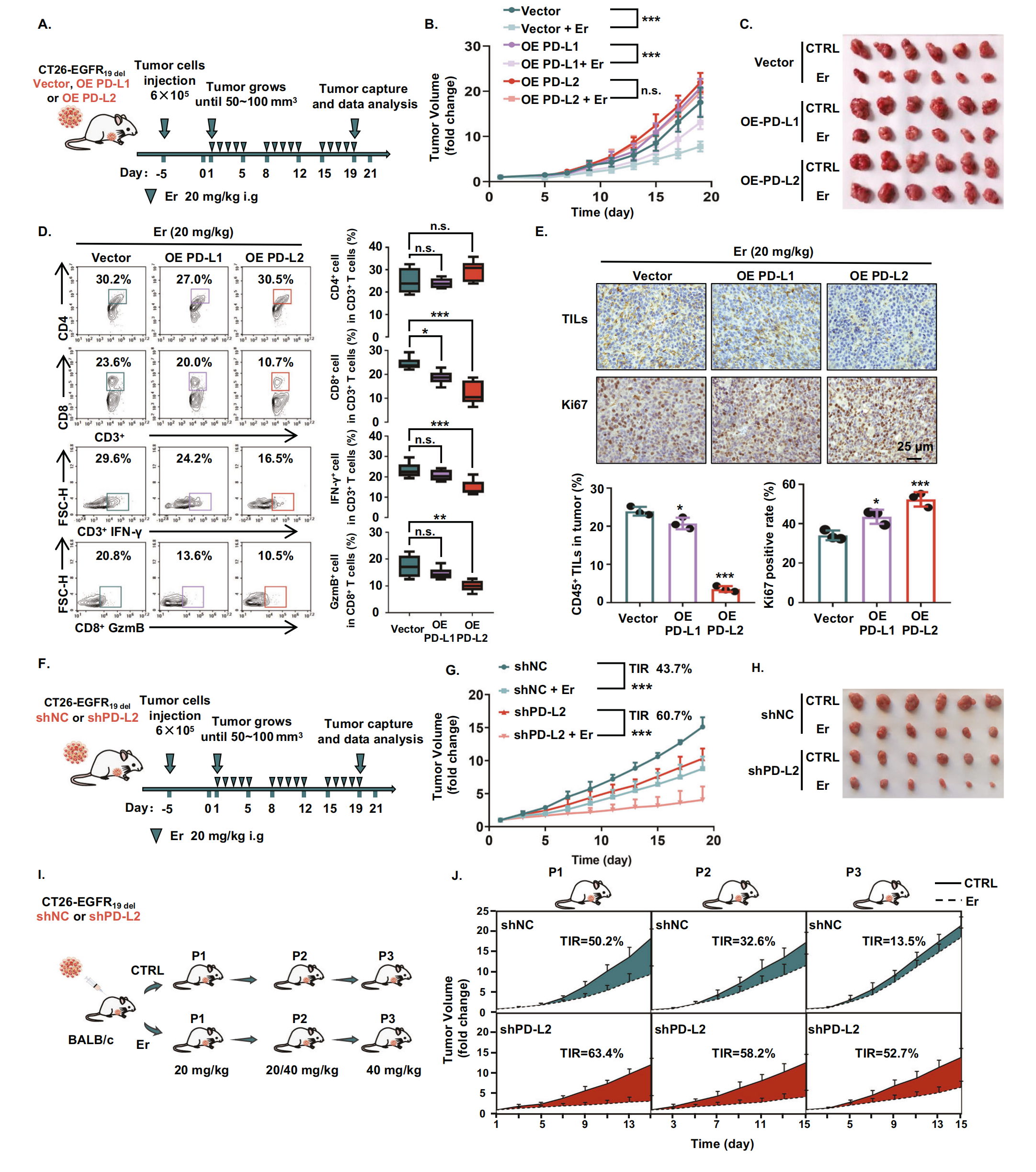
图4-PD-L2 通过影响肿瘤免疫环境在介导 EGFR-TKIs 耐药中发挥关键作用
鉴于 PD-L2 在体内调节 T 细胞活性中的关键作用,作者通过体内实验表明,PD-L2 的稳定沉默增加了 EGFR-TKI 的敏感性(图4F,G)。为了进一步研究 PD-L2 如何影响对 EGFR-TKI 的耐药动力学,作者在 EGFR-TKI 耐药同基因小鼠模型的开发中稳定地沉默了 CT26-EGFRmut 细胞系中的 PD-L2(图4H)。结果显示,厄洛替尼在第一代显著抑制 CT26-EGFR19del 衍生肿瘤的生长。重要的是,PD-L2 沉默导致对 EGFR-TKI 治疗的耐药性延迟发生。即使在第三代 (P3) 时对照组 (CT26-EGFR19del shNC 衍生肿瘤) 出现耐药性 (肿瘤抑制率 = 13.5%),PD-L2 沉默的肿瘤对 EGFR-TKIs 保持高敏感性 (肿瘤抑制率 = 52.7%) (图4I,J)。综上所述,作者的基因操作实验表明,PD-L2 通过影响肿瘤免疫微环境在介导 EGFR-TKIs 耐药中发挥关键作用。
- 阻断 PD-L2/PD-1 联合 EGFR-TKI 可抑制 EGFR-TKI 耐药细胞或肿瘤的生长
接下来,作者想进一步探讨阻断 EGFR-TKI 耐药细胞中的 PD-L2/PD-1 是否可以通过增强免疫反应来增强肿瘤细胞对 EGFR-TKIs 的敏感性。作者建立了携带对奥希替尼耐药的 LLC-EGFR19del 衍生肿瘤的同基因小鼠模型。小鼠接受生理盐水 (CTRL)、奥希替尼 (Os)、抗 PD-1 (αPD-1) 和奥希替尼 + 抗 PD-1 (Combi) 治疗(图5A)。作者的结果表明,与 CTRL 组相比,单独使用奥希替尼组小鼠的肿瘤体积和生存时间没有显着差异。PD-1 抑制剂对 EGFR-TKI 耐药肿瘤表现出部分抑制,并导致小鼠存活时间适度延长,尽管没有统计学意义。然而,奥希替尼和抗 PD-1 的组合显着抑制了肿瘤生长并延长了携带耐药肿瘤的小鼠的生存期(图5B,C)。以上结果表明,抑制 PD-1 与 EGFR-TKI 联合有效抑制了 EGFR-TKI 耐药肿瘤的生长。
此外,作者进一步确定了在耐药细胞中稳定敲低 PD-L2 是否可以增强这些细胞对 EGFR-TKIs 的敏感性,为了区分 PD-L1 和 PD-L2 的作用,作者生成了 PD-L1 和 PD-L2 稳定沉默的 PC9R 细胞系。与 PD-L1 沉默细胞相比,PD-L2 沉默的肿瘤细胞对 EGFR-TKI 更敏感,它们更有效地增加了 T 细胞的增殖和 CD8+ T 细胞的比例(图5D,E)。此外,作者使用抗体阻断 PD-L1、PD-L2 和 PD-1 的功能获得了一致的结论。抗 PD-L2 和 PD-1 的抗体使肿瘤细胞对 EGFR-TKI 更敏感,并增加了 T 细胞的增殖,CD8+ T 细胞的比例更高(图5F, G)。上述体内和体外实验表明,PD-L2 的高表达通过影响免疫反应来影响 EGFR 突变细胞对 EGFR-TKIs 的敏感性。因此,PD-1/PD-L2 信号传导,而不是 PD-1/PD-L1 信号传导,可能在介导对 EGFR-TKI 的耐药中发挥重要作用。
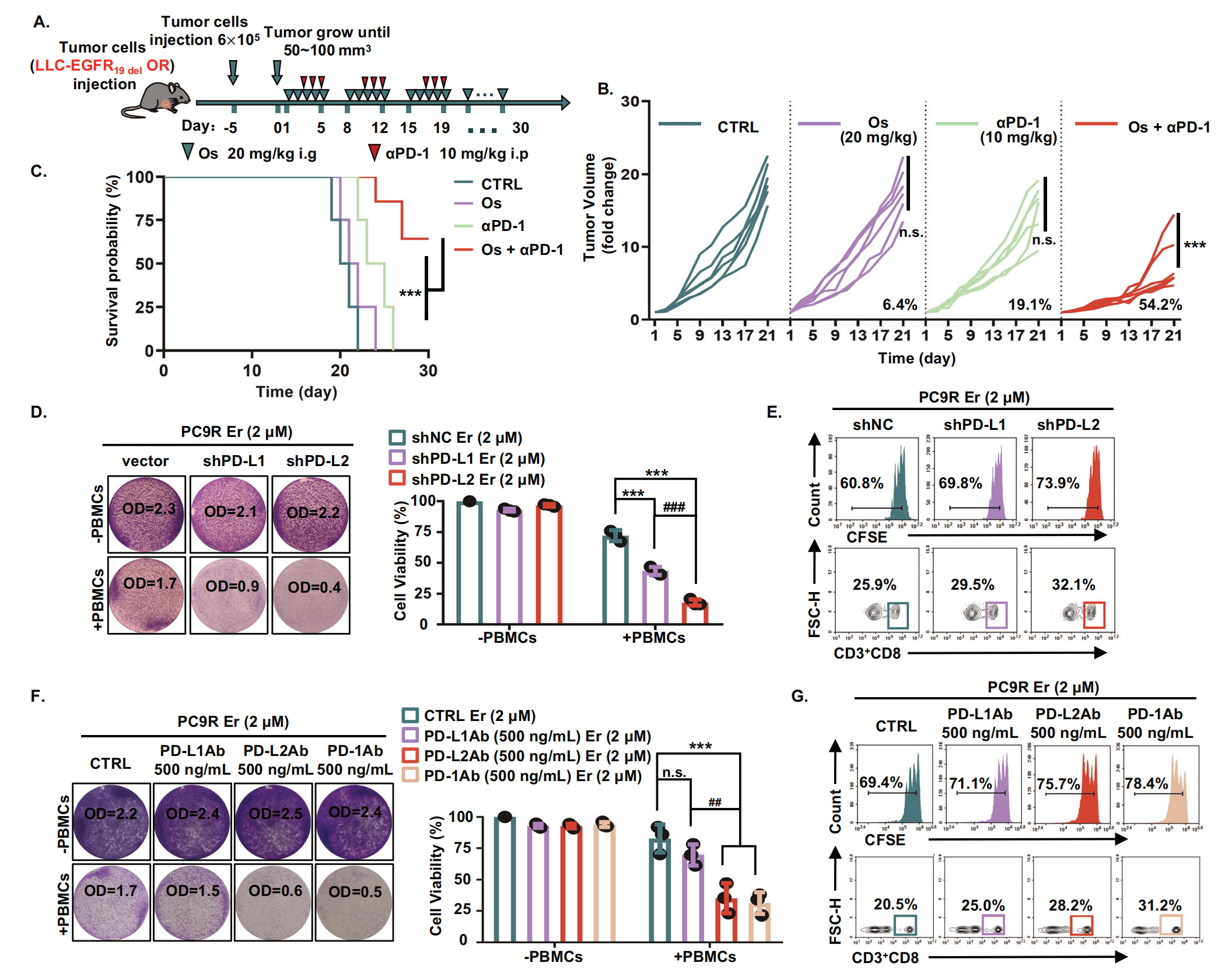
图5-阻断 PD-L2/PD-1 联合 EGFR-TKI 可抑制 EGFR-TKI 耐药细胞或肿瘤的生长
- PD-L2 过表达抑制肿瘤细胞凋亡并促进对 EGFR-TKI 的耐药
接下来,作者进一步探讨了 PD-L2 表达升高影响免疫反应以降低肿瘤对 EGFR-TKIs 敏感性的机制。作者获得了来自空载体或 PD-L2 过表达的同基因小鼠中 CT26-EGFR19del 细胞的肿瘤组织的单细胞转录组谱。作者对来自空载体样本和 PD-L2 过表达样本的整合单细胞数据集进行了聚类分析,并确定了六个主要的不同细胞群(图6A、B)。作者发现 PD-L2 过表达组中癌细胞的数量显着增加,T 细胞的比例显着降低(图6C)。为了探索 EGFR-TKIs 耐药的机制,作者分析了癌细胞的转录组特征。与空载体组相比,对 PD-L2 过表达组的差异表达基因进行 KEGG 富集分析。作者观察到细胞凋亡通路显著富集(图 6D)。GSEA 分析显示,PD-L2 过表达组癌细胞中与细胞凋亡途径相关的基因显著下调(图6E)。这些结果表明,EGFR 突变肿瘤细胞中 PD-L2 表达的增加可导致细胞凋亡减少。作者的体内实验数据进一步验证了这些结论。TUNEL 染色显示,与对照组相比,TKI 处理后 CT26-EGFR19del PD-L2 过表达组肿瘤细胞凋亡受到显著抑制(图6F)。基于这些结果,作者假设 EGFR 突变的肿瘤细胞中 PD-L2 表达增加会抑制癌细胞的凋亡,从而导致癌细胞的持续生长,耐药性增加。
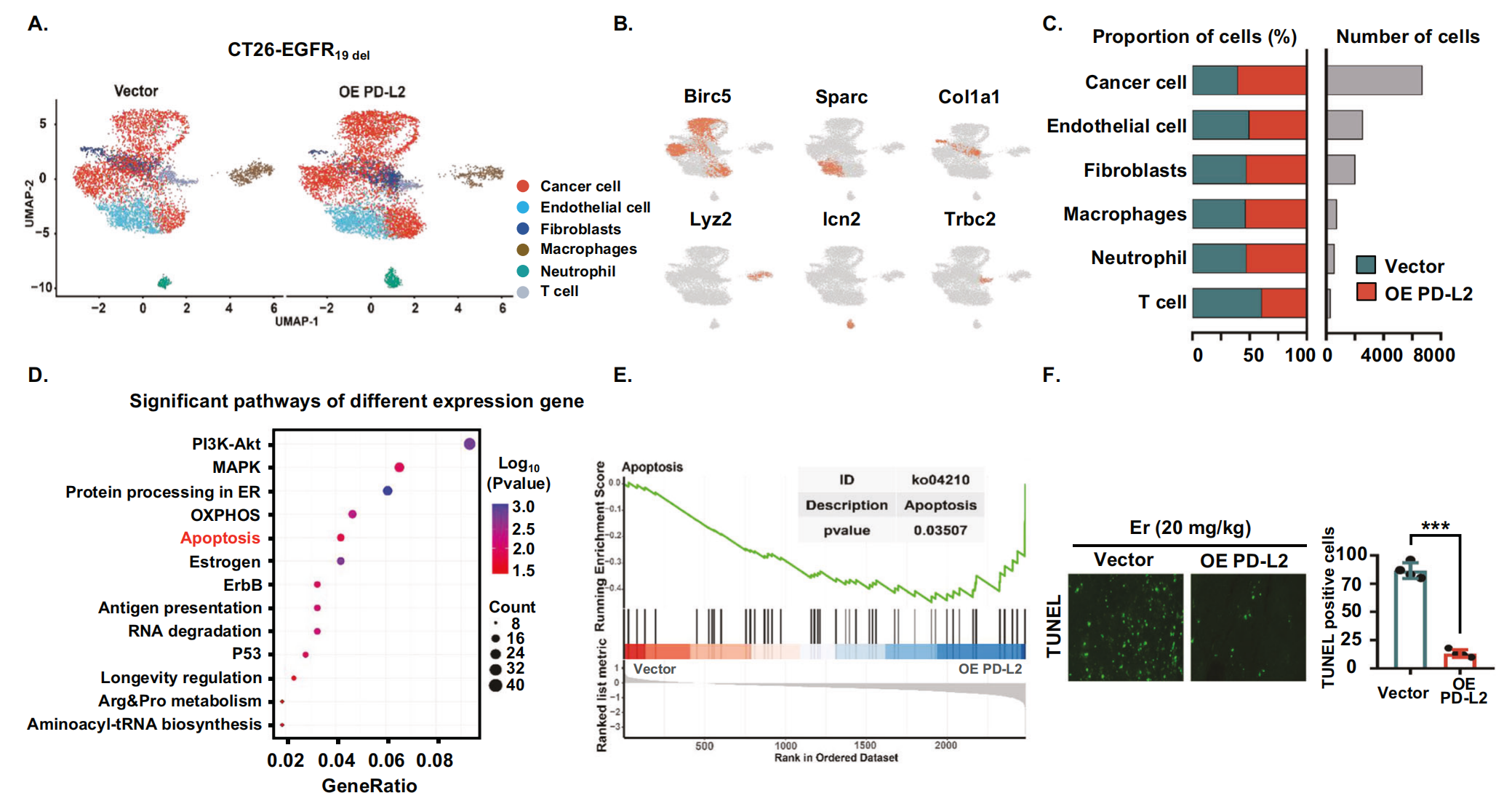
图6-PD-L2 过表达抑制肿瘤细胞凋亡并促进对 EGFR-TKI 的耐药
- PD-L2 过表达抑制 CD8+T 细胞通过穿孔素-颗粒酶 B 通路诱导肿瘤细胞凋亡的功能
为了进一步了解 PD-L2 对肿瘤免疫环境的抑制作用,作者回到了作者的单细胞测序数据。根据 CD45 的表达,将空载体和 PD-L2 过表达组的肿瘤环境中细胞分为 CD45- 肿瘤细胞和 CD45+ 免疫细胞(图7A)。然后根据已知遗传标记的表达和细胞亚群的特异性注释对 CD45+ 免疫细胞进行分组(图 7B)。在 PD-L2 过表达组中,作者观察到 CD8+ T 细胞群的大小显着减小(图7B,C)。因此,作者通过 KEGG 富集分析了空载体组和 PD-L2 过表达组 CD8+ T 细胞中的差异表达基因(图 7D)。作者发现与增殖和细胞毒功能相关的通路显著富集(图7E)。为了进一步证实 PD-L2 过表达显着影响 CD8+ T 细胞活性,作者从总 T 细胞中分离 CD8+ T 细胞,并将它们与 PD-L1 或 PD-L2 基因操纵的小鼠肿瘤细胞共培养。结果显示,CD8+ T 细胞的增殖在 PD-L2 过表达细胞中受到显著抑制,而在 PD-L1 过表达细胞中则不受抑制(图7F)。然后,作者从总 T 细胞中分离 CD4+ T 细胞,并将它们与 PD-L1 或 PD-L2 基因操纵的小鼠肿瘤细胞共培养。值得注意的是,PD-L2 过表达对 CD4+ T 细胞的增殖没有影响,而 PD-L1 过表达显著抑制了 CD4+ T 细胞的增殖(图7G)。这表明 PD-L2 和 PD-L1 介导 T 细胞抑制的分子机制存在差异。
许多研究表明,CD8+ T 细胞可通过 Fas-FasL 通路或穿孔素-颗粒酶 B 通路诱导肿瘤细胞凋亡。因此,作者接下来使用纯化的 CD8+ T 细胞与 PD-L1 或 PD-L2 基因操纵的小鼠肿瘤细胞共培养,并研究 EGFR-TKIs 处理后肿瘤细胞的凋亡情况。结果显示,与 PD-L1 过表达细胞相比,PD-L2 过表达减少了 EGFR-TKI 处理后 CD8+ T 细胞介导的肿瘤细胞凋亡(图7H)。接下来,作者试图检查肿瘤细胞 PD-L1 或 PD-L2 过表达对 CD8+ T 细胞分泌细胞毒性细胞因子的影响。正如预期的那样,与 PD-L1 过表达相比,PD-L2 过表达显着抑制了 CD8+ T 细胞颗粒酶 B 和穿孔素伴随 EGFR-TKI 治疗的分泌(图7I)。综上所述,EGFR 突变肿瘤细胞中 PD-L2 的过表达显著抑制了 CD8+ T 细胞的增殖和细胞毒功能,主要是通过抑制穿孔素和颗粒酶 B 的分泌,从而减少肿瘤细胞的凋亡。
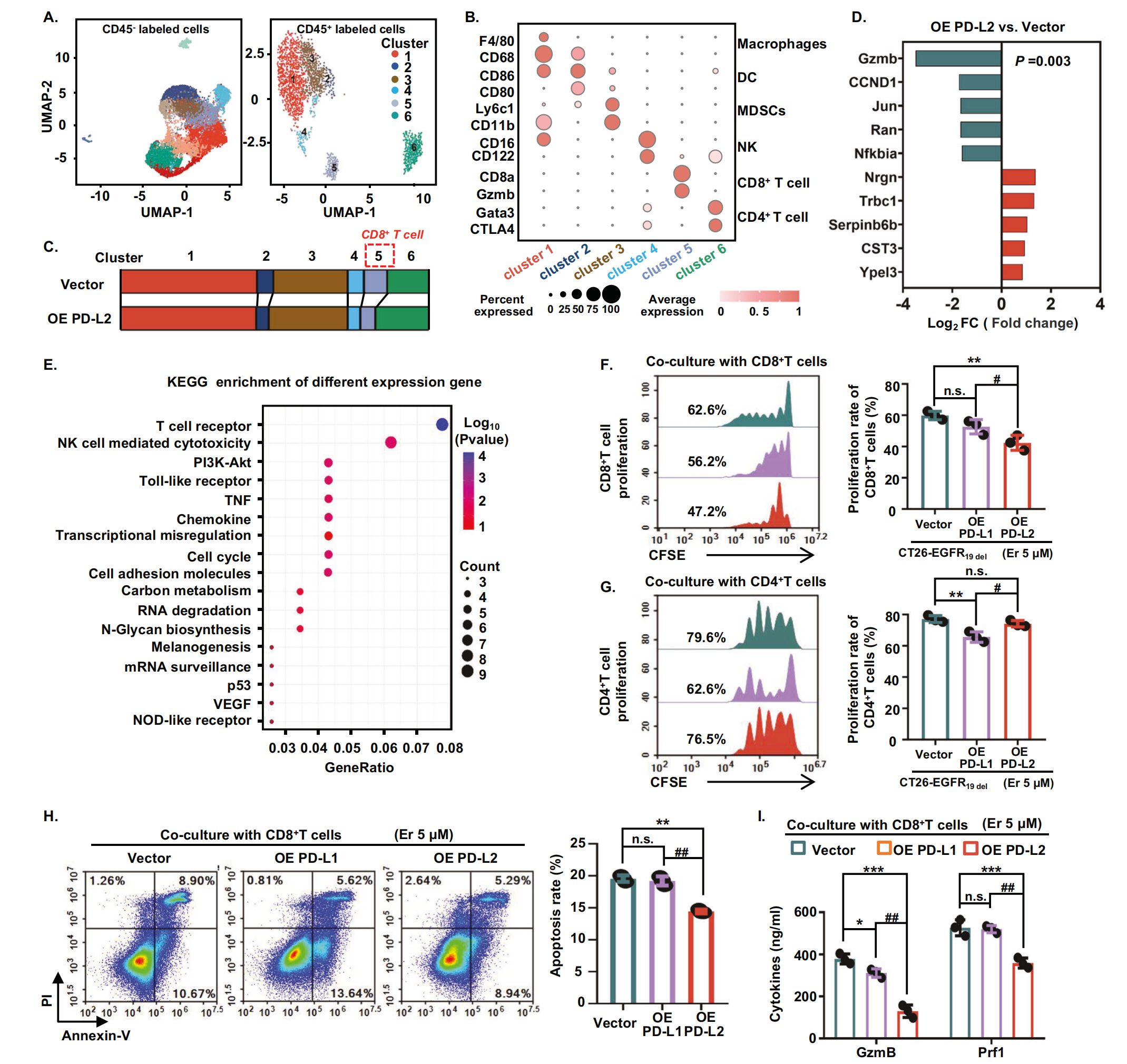
图7-PD-L2 过表达抑制 CD8+ T 细胞通过穿孔素-颗粒酶 B 通路诱导肿瘤细胞凋亡的功能
- ZU 作为天然存在的 PD-L2 小分子抑制剂的筛选和活性评价
由于目前市场上没有靶向 PD-L2 的药物,作者使用 HTRF 技术筛选了 1300 多种天然小分子化合物的库,以鉴定 PD-L2 的特异性抑制剂。作者发现了一种小分子化合物十一烯酸锌 (ZU),它选择性地抑制 PD-L2/PD-1 结合,EC50 8.4 μM,而 PD-L1/PD-1 为 >80 μM(图8A, B)。ZU 是由蓖麻油通过一系列化学反应和净化过程生产的。CETSA 实验表明,与 PD-L1/PD-1 相比,ZU 有效抑制了 PD-L2 与 PD-1 的结合(图8C)。这些结果表明 ZU 具有阻断 PD-L2/PD-1 相互作用的特异性能力。此外,作者利用空载体或 PD-L2 过表达的小鼠 CT26 细胞来验证 ZU 的体外抗肿瘤作用。在 40 μM 浓度下,ZU 不会显著降低 CT26 空载体和 PD-L2 过表达细胞的活力(图8D),这表明 ZU 没有直接的细胞毒性作用。然而,当肿瘤细胞与活化的 T 细胞共培养时,与 CT26 空载体细胞相比,ZU (40 μM) 显着抑制了 CT26 PD-L2 过表达细胞的活力(图8D)。这表明 ZU 通过靶向 PD-L2/PD-1 的免疫调节发挥抗肿瘤作用。使用具有空载体和 PD-L2 过表达的 PC9 细胞获得了类似的结果(图8D)。此外,作者研究了 ZU 和 EGFR-TKI 对亲本和 TKI 耐药 NSCLC 细胞的联合抑制作用。对于耐药细胞,结果表明,厄洛替尼联合 ZU 对 827 R 或 PC9R 细胞的生长抑制具有协同作用,与单独使用厄洛替尼相比,耐药细胞的存活率显著降低(图8E)。重要的是,在没有活化的 T 细胞的情况下,厄洛替尼联合 ZU 的细胞杀伤效果没有显着差异(图8E)。上述结果进一步表明,靶向 PD-L2 的 ZU 与 EGFR-TKI 的组合对耐药肿瘤表现出协同抑制作用。
接下来,作者评估了 ZU 是否通过靶向 PD-L2 可以改善肿瘤免疫反应。流式细胞术分析显示,与单独使用厄洛替尼或 ZU 相比,厄洛替尼联合 ZU 显着增加了 CD8+ T 细胞的比例和颗粒酶 B 的表达(图8F,G)。作者进一步检测到与活化 T 细胞共培养的 827 个 R 或 PC9R 细胞的凋亡水平。作者发现厄洛替尼联合 ZU 显着促进免疫细胞介导的 EGFR-TKI 耐药细胞凋亡(图8H)。相比之下,当用厄洛替尼加 ZU 处理耐药细胞时,在没有活化的 T 细胞或细胞凋亡抑制剂(泛半胱天冬酶抑制剂 Z-VAD-FMK 和特异性颗粒酶 B 诱导的细胞凋亡抑制剂 Z-IETD-FMK)的情况下未观察到细胞凋亡(图8H)。这进一步证明了耐药细胞的凋亡是由免疫细胞介导的。此外,颗粒酶 B (Z-IETD-FMK) 的抑制有效地消除了细胞凋亡,突出了颗粒酶 B 在 ZU 和 EGFR-TKI 诱导的耐药癌细胞凋亡中的关键作用(图8H)。总之,这些结果表明天然小分子抑制剂 ZU 可以有效和特异性地抑制 PD-L2。当与 EGFR-TKIs 联合使用时,ZU 可以通过在体外激活免疫细胞介导肿瘤细胞凋亡来显着逆转肿瘤细胞的耐药性。
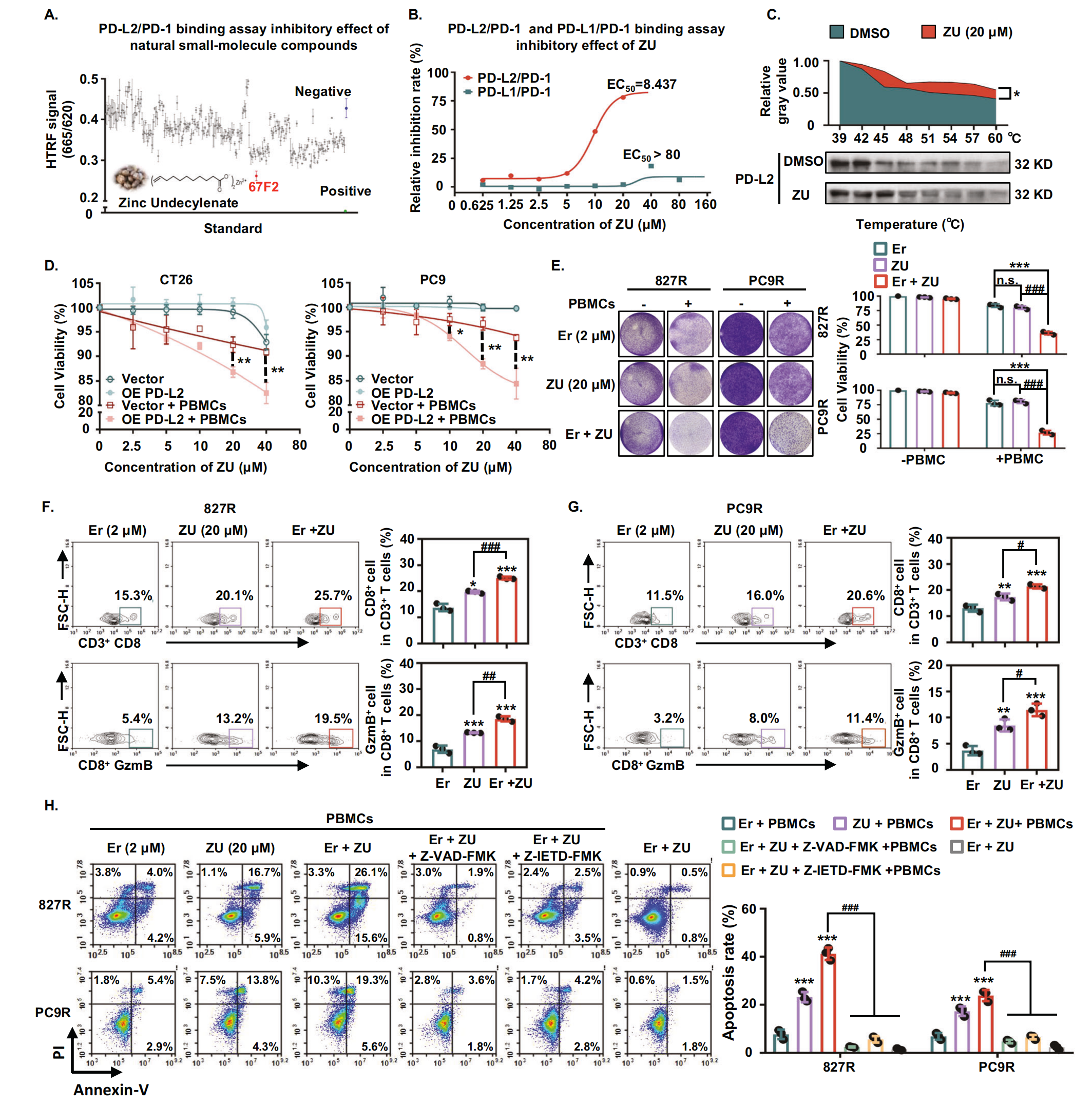
图8-ZU 作为天然存在的 PD-L2 小分子抑制剂的筛选和活性评价
- ZU 改善抑制性肿瘤免疫环境,在体内逆转 EGFR-TKIs 耐药
为了进一步证实 ZU 和 EGFR-TKIs 的组合协同改善肿瘤免疫环境并逆转肿瘤耐药性,作者评估了 ZU + 厄洛替尼在携带 PD-L2 过表达的 CT26-EGFR19del 来源的肿瘤的 BALB/c 同基因小鼠中的疗效(图9A)。肿瘤体积曲线显示,与盐水处理组相比,单独使用厄洛替尼在治疗 15 天后表现出边缘性肿瘤生长抑制(图9B),这表明 PD-L2 的过表达增加了肿瘤细胞对 TKI 的耐药性。ZU 单次治疗并未阻断肿瘤生长(图9B),这表明单独靶向 PD-L2 不能逆转厄洛替尼耐药性。正如预期的那样,阳性对照组 (Er + ADVshPD-L2) 在所有测试的模型中反应最好(图9B)。此外,ZU + 厄洛替尼组与阳性对照组相比没有显著差异,这表明 ZU 可以有效抑制 PD-L2,并且当与厄洛替尼联合使用时,可以显着逆转厄洛替尼耐药性(图9B)。此外,流式细胞术和免疫组化结果显示,联合治疗增加了肿瘤组织中浸润白细胞(CD45+)的比例、T 细胞的活化 (CD3+CD69+)以及CD8+ T 细胞(CD8+GzmB+)的比例和功能(图9C)。免疫组化显示,联合治疗显著抑制了增殖(图9D)。为了全面评价 EGFR-TKIs 联合 ZU 的疗效,作者建立了携带对奥希替尼耐药的 LLC-EGFR19del 衍生肿瘤和对厄洛替尼耐药的 CT26 EGFR19del 衍生肿瘤的同基因小鼠模型。小鼠接受生理盐水(CTRL)、单独 EGFR-TKIs、 单独 ZU和EGFR-TKIs + ZU处理。作者的结果表明,与生理盐水组相比,单独使用 EGFR-TKIs 组或单独使用 ZU 组小鼠的生存时间没有显着差异(图9E, F)。然而,ZU 和 EGFR-TKIs(奥希替尼或厄洛替尼)的组合显着延长了携带耐药肿瘤的小鼠的生存期(图9E,F)。这些结果表明,ZU 联合 EGFR-TKIs 可以通过改善同基因小鼠模型中的肿瘤免疫环境来有效逆转对 EGFR-TKIs 的耐药性。综上所述,ZU 通过抑制 PD-L2 增强 CD8+ T 细胞的功能,其与 EGFR-TKIs 的联合作用可有效抑制耐药肿瘤的增殖。
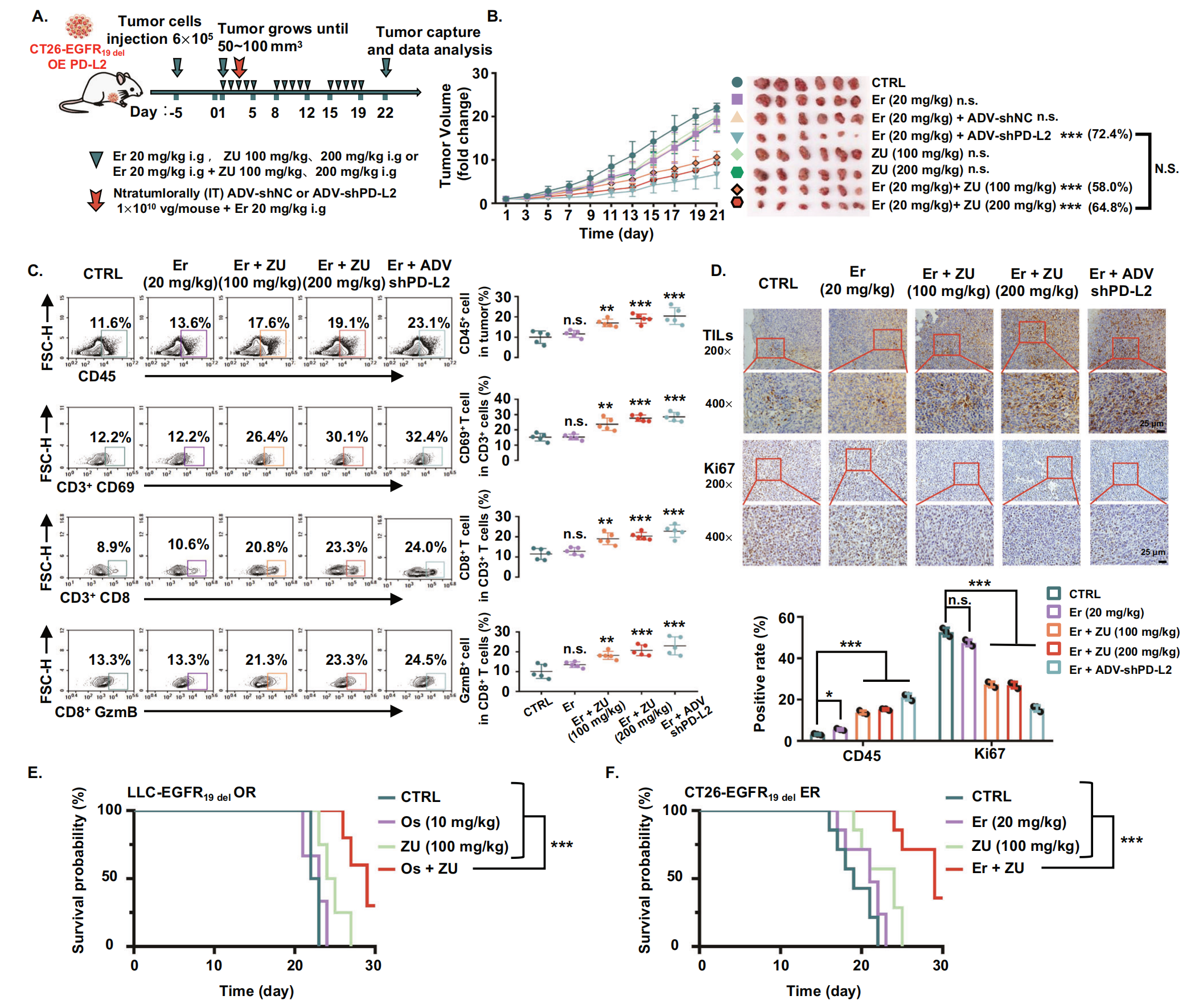
图9-ZU 改善抑制性肿瘤免疫环境,在体内逆转 EGFR-TKIs 耐药
研究总结
在该研究中,由于难以获得对 EGFR-TKIs 耐药的临床肿瘤样本,作者使用转染人 EGFR 突变体 (外显子19 缺失) 的小鼠细胞系建立 EGFR-TKI 耐药同基因小鼠模型约五个月,这是使用基因工程小鼠难以实现的。值得注意的是,该研究的 TKI 耐药模型的关键点是将初始抑制率与不同传代模型中的抑制率进行了比较,例如 Os 治疗的 LLC 从 44% 到 -12.3%,Er 治疗的 CT26 从 55% 到 11.2%。因此,该研究认为该模型可以反映临床 TKI 给药中的耐药过程。这种方法使作者能够研究 EGFR-TKIs 治疗过程与肿瘤免疫环境之间的关系。作者的结果表明,EGFR-TKIs 对肿瘤免疫环境具有显着而显着的影响。当 EGFR-TKIs 治疗有效时,抗肿瘤免疫反应增强。然而,当发生耐药性时,作者观察到肿瘤浸润白细胞的比例显着降低,CD8+ T 细胞的比例和功能降低。不断变化的肿瘤免疫环境可能是 PD-1 阻断治疗后立即再次用 EGFR-TKIs 攻击对 EGFR-TKI 耐药的 NSCLC 患者有效的原因 。此外,作者得出结论,抑制性肿瘤免疫环境是参与对 EGFR-TKIs 耐药的机制之一。因此,作者从非遗传角度揭示了 EGFR-TKIs 耐药的新机制。




 京公网安备 11011302003368号
京公网安备 11011302003368号