


01 研究背景
荔枝是一种重要的风味独特的热带水果,具有极高的营养价值和经济价值。荔枝品种按果实成熟期可分为极早熟品种(EEMC)、中早熟品种(EMC)和晚熟品种(LMC)3大类;EEMCs品种稀少,生产价值不高,果实品质较好的品种多属于LMC品种;目前不同成熟期品种间的差异研究较少。荔枝目前在我国的云南、海南和广西、广东等地均有栽培种植,然而,确切的起源中心和荔枝驯化的历史仍然是未知的。研究荔枝基因组的结构和进化对促进荔枝及无患子科近缘植株的遗传改良具重要价值。
02 材料与方法
基因组:荔枝栽培种“Feizixiao”;~184x Illumina+~58.6x PacBio CLR+~144x Hi-C
重测序:38份野生种和34份栽培种;~13x
03 研究结果
1.“Feizixiao”荔枝基因组组装注释
利用三代PacBio、二代illumina测序技术以及Hi-C技术测序,通过优化基因组组装策略,获得高质量的“妃子笑”荔枝基因组染色体水平的组装,主要含15条假染色体序列,大小470Mb,杂合度2.27%,BUSCO评估显示基因组组装完整性为96.2%;同时完成基因组编码基因的结构注释,共注释得到31896个结构基因,注释完整度BUSCO评估94.8%,充分表明所获得的“妃子笑”荔枝基因组质量非常高。荔枝基因密码的破译将为未来荔枝功能基因组研究提供重要的参考。
比较基因组分析表明,荔枝和龙眼-10百万年前(mya)从祖先物种中分离开来,而荔枝所在的无患子科与芸香科大约在67.6mya分离开。荔枝、龙眼、文冠果和甜橙自共享y三倍化事件后并无独立的WGD事件。
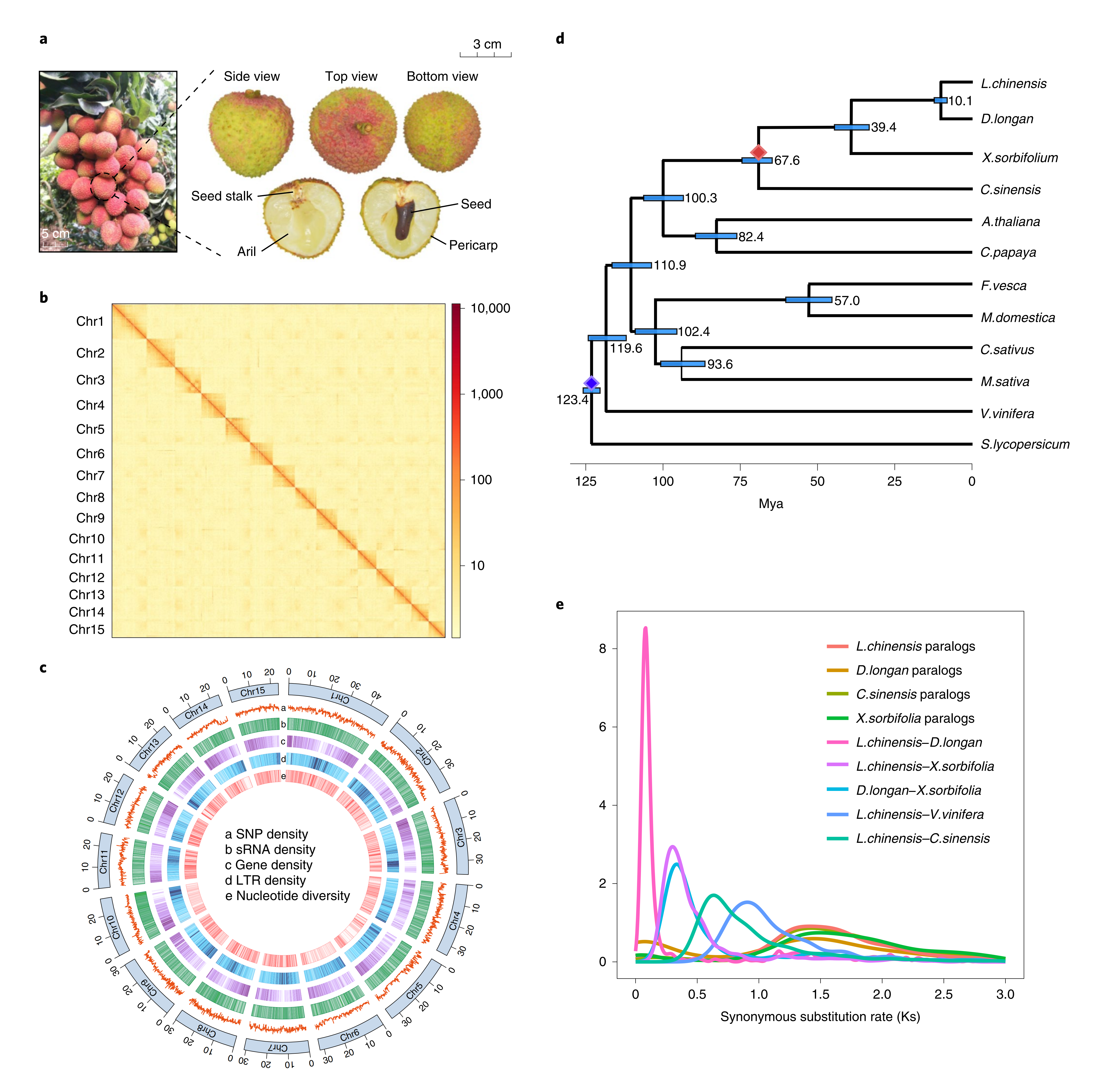
图1-荔枝基因组的组装、组成和进化
2.荔枝的起源与驯化
利用通过对72份荔枝进行重测序分析,发现荔枝大致可分为3个群体,即云南极早熟品种(EEMC/YNW)、海南晚熟品种(LMCHNW)、早熟栽培种(EMC)。其中值得注意的是,大新野生和博白野生荔枝虽然都位于广西,但两者间却存在明显的遗传差异。分析发现云南野生种群经历了强烈的驯化瓶颈。
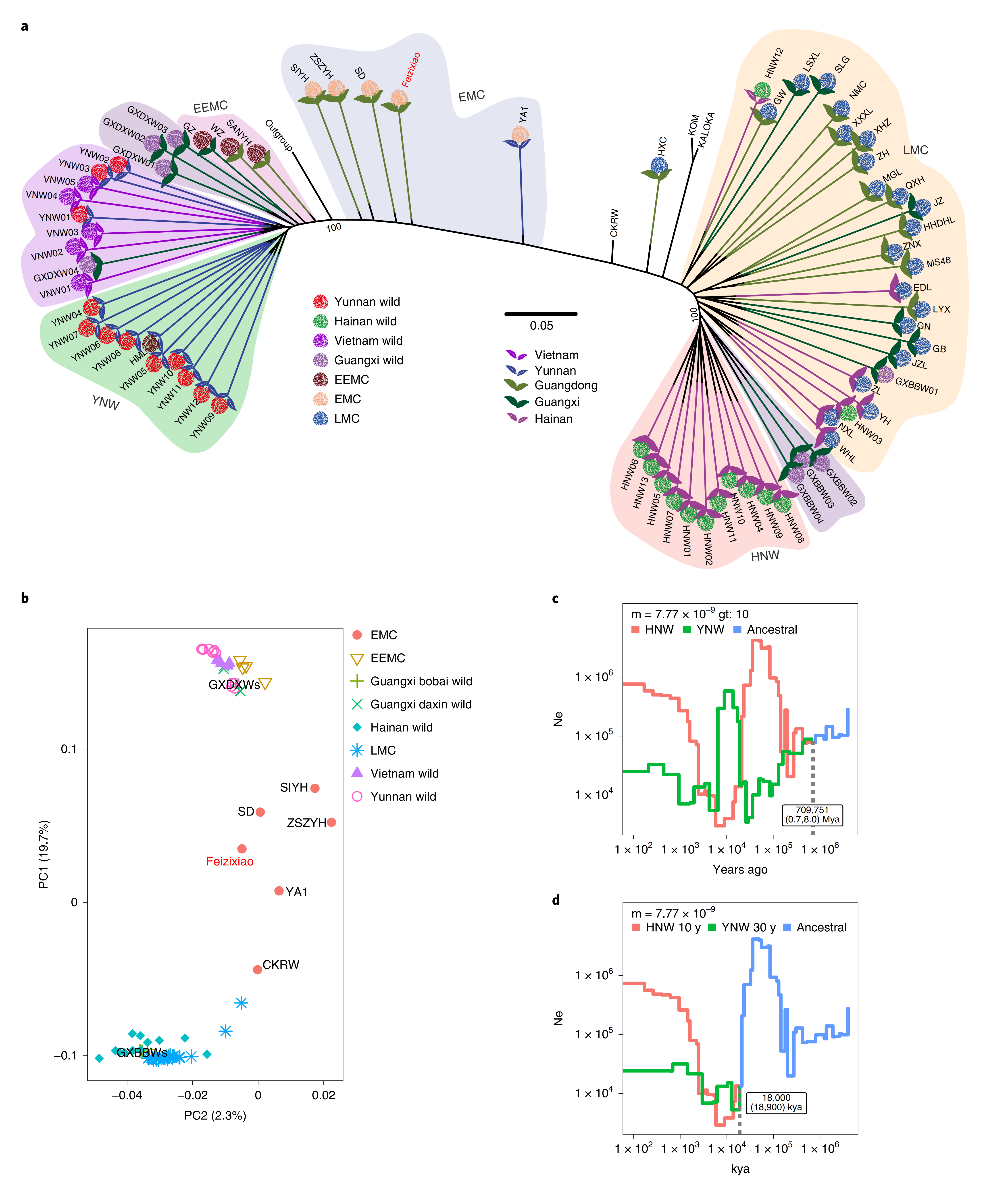
图2-荔枝重测序群体分析
3.荔枝单倍型注释和扥给基因差异表达
作者通过SNP分型和单细胞10x Gemomics测序,利用“妃子笑”荔枝的高杂合性(2.27%),成功获得的两个单倍型的基因组(云南单倍型,HY;海南单倍型,HH)。对39个妃子笑”不同组织和时期样本进行转录组测序分析,共鉴定到13517个等位差异表达基因(DEA)。DEA在基因组上的分布并不是均匀的,在某些区域具有集中现象,可能与特定的生物学过程相关。还发现,DEA和等位等量表达基因(EEA)具有相同的Ks值,但是DEA经受更强的选择压力。
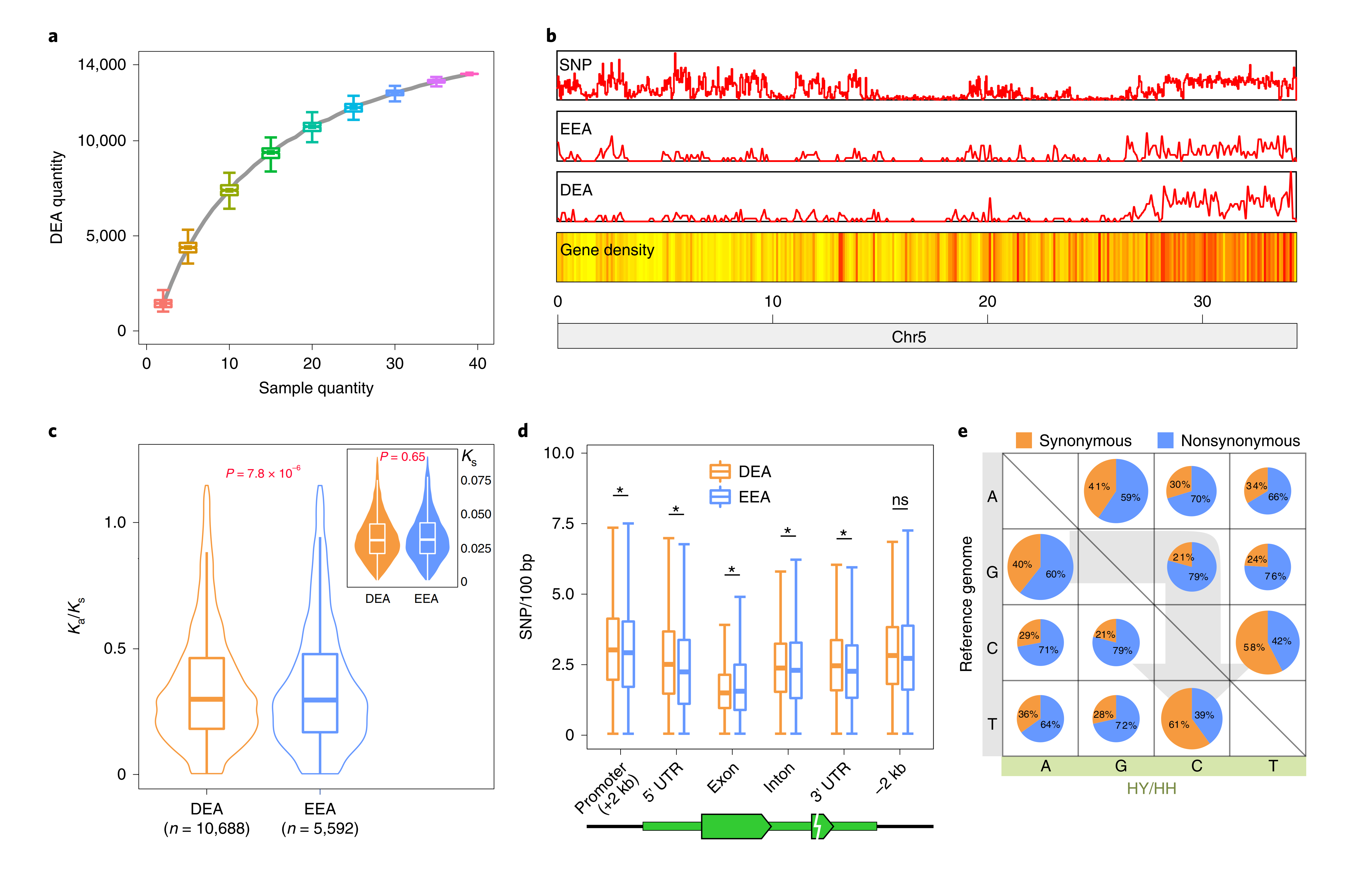
图3-荔枝等位基因差异表达
4.COL307基因调控果实成熟
为了进一步剖析荔枝果实成熟的调控网络,作者对72个样本进行GWAS分析,20个显著性位点的±50kb内含有109个基因,与正选择基因和开花相关基因取交集,最终筛选到目标基因COL307。COL307下游含有3.7kb序列的缺失,进一步分析发现极早熟果实品种中为纯合缺失,晚熟品种中为纯合不缺失,早熟品种中为杂合缺失,该缺失与果实成熟期高度连锁,可以开发为简便的分子标记用于荔枝辅助育种。
04 研究总结
基于荔枝(Litchi chinensis)基因组的高度杂合(~2.27%),构建了两套高质量妃子笑”单倍型基因组(470 Mb)。72份荔枝重测序分析发现,来自云南的一个野生群体的极早熟荔枝品种主要与“妃子笑”的其中一个单倍型对应;来自海南的一个野生群体的晚熟荔枝品种则与另外一个单倍型对应。早熟荔枝品种可能是极早熟荔枝品种与晚熟荔枝品种的杂交形成的,在广东培育而成。包含一对CO-likc基因的3.7kb序列的缺失变异可能调控着不同荔枝品种间的果实成熟差异。



 京公网安备 11011302003368号
京公网安备 11011302003368号