

2024年4月22日,浙江省农业科学院李国景研究员团队与浙江大学张明方教授团队合作的豇豆基因组研究成果以“Differential selection of yield and quality traits has shaped genomic signatures of cowpea domestication and improvement”为题,在国际遗传学期刊Nature Genetics(IF:30.8)上在线发表,百迈客生物参与了其中的测序和部分分析工作。文章通过三代PacBio HiFi数据成功构建了1个粮用豇豆和1个菜用豇豆的高质量基因组,并对全世界收集的344份核心种质进行重测序,揭示了豇豆驯化和改良的基因组选择印迹,阐明了豇豆产量和品质协同提升的遗传机制。

研究背景
豇豆(Vigna unguiculata L. Walp., 2n = 2x = 22)起源于非洲,在世界范围内作为粮食、蔬菜或牲畜饲料种植。驯化豇豆已形成两个主要栽培亚种:非洲的粮用豇豆和亚洲的菜用豇豆。这两个亚种在许多重要的农艺性状上差异很大,如荚长(PL)、单荚粒数(GNP)和营养品质等。
之前的研究已经报道了一些控制豇豆驯化与改良性状的QTLs,如裂荚性(PS)、PL、荚果品质和种子大小等。然而,与亚种驯化相关的全基因组选择特征尚不清楚。尽管高质量的粮用豇豆基因组已经发布,但由于高质量菜用豇豆基因组信息的缺乏,阐明不同豇豆亚种驯化和改良的遗传机制仍存在严重的阻碍。因此,全面解析豇豆基因组可以为不同亚种的驯化与改良提供见解,并为其育种改良提供重要的基因组资源。
材料方法
Denovo:
菜用豇豆G98,Illumina:107.99x;PacBio HiFi:49.36x;Hi-C:73.88Gb
粮用豇豆G323,Illumina,121.57x;PacBio HiFi:41.82x;Hi-C:68.64Gb
重测序:
344份全世界收集的豇豆核心种质,其中包括342份栽培豇豆(87份粮用豇豆、244份菜用豇豆和11份未知用途豇豆)和2份野生豇豆;Illumina测序,10x深度
基因单倍型验证:
菜用豇豆地方品种 ‘ZN016’ 和菜用豇豆育成品种‘Zhijiang282构建的RIL群体(183 lines)
G98和G323构建的F2群体(165 individuals)
研究结果
- 豇豆基因组的构建
该研究选择了长荚菜用豇豆(G98)和抗病性强的粮用豇豆(G323)进行基因组Denovo。K-mer预估G98和G323的基因组大小分别为623.16Mb和597.42Mb,最终组装的基因组大小分别为568.24Mb(scaffold N50 = 49.41Mb)和552.66Mb(scaffold N50 = 49.35Mb)。二代回比率(G98:99.19%;G323:99.69%)、CEGMA(98%)、BUSCO(95%)和Merquery(G98:44.48;G323:47.17)评估表明本次构建了2个高质量的染色体水平豇豆基因组。
G98和G323分别预测到33,159和33,222个蛋白编码基因,重复序列占基因组的56.97%(G98)和55.25%(G323),以Gypsy的长末端重复反转录转座子(LTRs)为主,分别占基因组的17.19%(G98)和18.72(G323)。
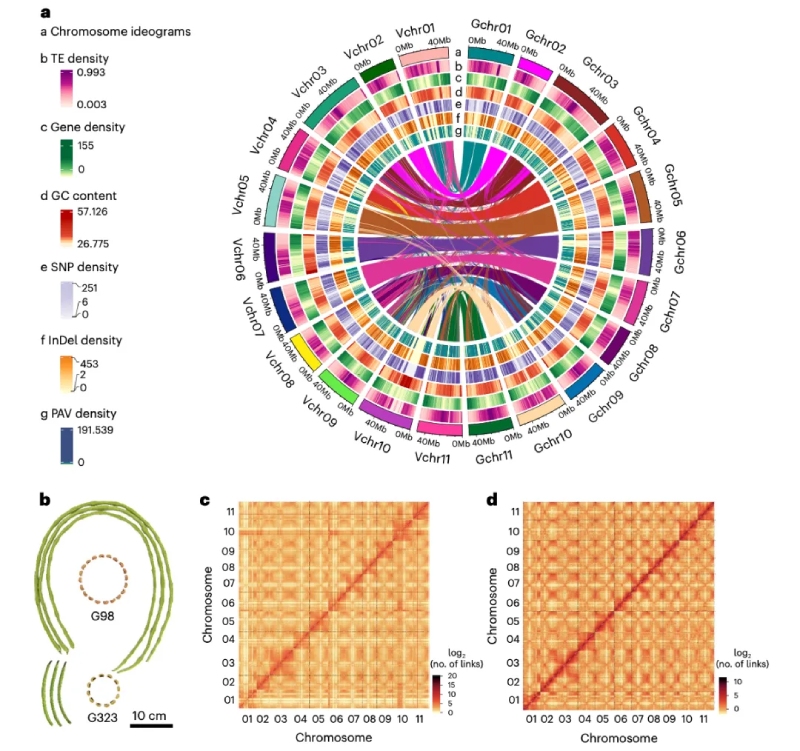
图1-菜用豇豆(G98)和粮用豇豆(G323)的高质量基因组组装
- 豇豆的系统进化与SVs
对25个物种进行系统进化树分析显示豇豆与赤小豆(V. angularis)和绿豆(V. radiata)亲缘关系较近,它们的分化时间大约在6-27百万年前(图2a),这与之前的报道一致。基因家族分析发现G98中有512个家族发生了显著扩张,这些扩张的基因家族在参与膜组织的鞘糖脂代谢途径显著富集,可能与菜用豇豆的豆荚发育相关。相反,G323扩张基因在氨基酸糖和核苷酸糖代谢、硫代葡萄糖苷-谷氨酰水解酶和半乳糖代谢等能量产生和转化途径中显著富集(图2b, c),这可能与粮用豇豆更高的碳水化合物积累和防御反应有关。
作者以G323为参考基因组,对2个基因组进行变异检测,在G98上共发现2,219,947个SNPs,其中38,420个SNPs可能引起基因功能的改变;发现407,119个InDels,其中62.50%的可能引起蛋白编码的改变;另外,发现13,541个SVs(包括963个TRANS,74个INVS,3,701个DUPs,7,112个PAVs和1,691个CNVs)。
值得注意的是,作者发现了5个较大的SV区域(> Mb)(图2d, f)。Chr1包含一个7.5 Mb的INV和多个相邻的TRANS,分别包含61个和31个基因(图2d)。Chr6包含一个4.73 Mb的INV区和一个5.14 Mb的INV区,包含两个INVs,两个DUPs和两个TRANS。这两个区域分别包含42和52个基因(图2e)。Chr10含有最大的一个INV,共包含224个基因(图2f)。在其余染色体上共检测到13个其它SV区域。
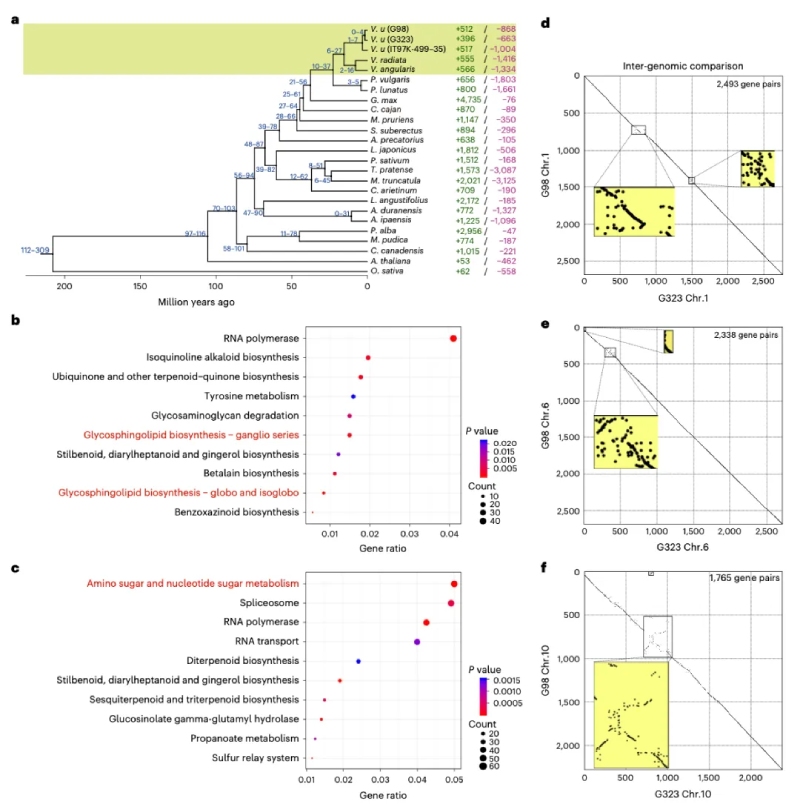
图2-豇豆系统发育和基因组结构变异分析
- 豇豆亚种的种群结构与分化
作者进一步对全世界收集的344份豇豆核心种质进行重测序,PCA分析将其划分为2个聚类(图3a),主要由粮用豇豆(cluster I)和菜用豇豆(cluster II)组成。利用菜豆作为外群,系统发育树将其划分为以粮用豇豆(G)、菜用豇豆地方品种(VL)和菜用豇豆育成品种(VC)为中心的3个类群(图3b)。群体结构分析也支持了PCA和系统发育树的结果(图3b)。
对三个亚群的核苷酸多样性(π)和群体差异(Fst)进行分析(图3c)。G组(π = 0.0007)核苷酸多样性显著高于VL组(π = 0.00047)和VC组(π = 0.00024)。G-VC(0.1903)和G- VL(0.0924)的FST值均高于VC-VL(0.0498)。此外,与VL和VC组相比,该组的连锁不平衡衰减更快,表明粮用豇豆的遗传重组程度更高(图3d)。
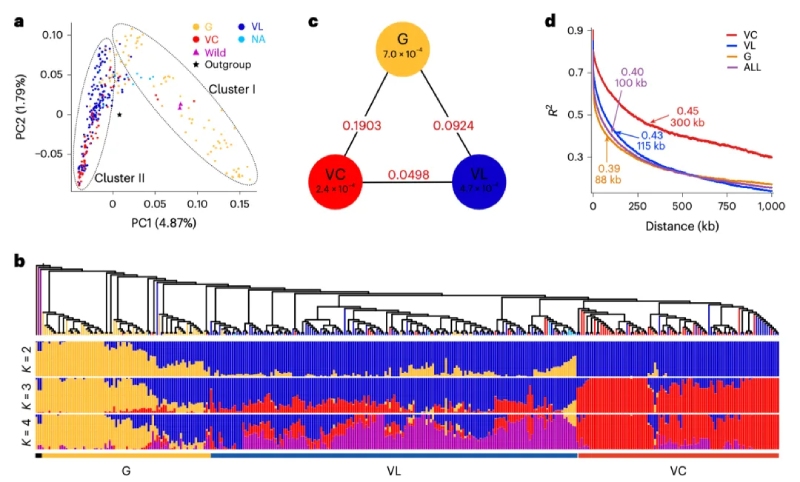
图3-344份豇豆材料群体结构和遗传多样性
- 豇豆驯化与改良的基因组特征
为了研究自然或人工选择对豇豆分化的影响,作者通过选择清除分析比较了三个豇豆亚群之间的基因组选择特征,鉴定出239个与豇豆驯化和改良相关的基因。
进一步结合GWAS分析,共鉴定出与嫩荚总淀粉含量(PTS)、籽粒总淀粉含量(STS)、单荚籽粒数(GNP)、嫩荚可溶性糖含量(PSS)和荚长(PL)相关的18个受选择的区域。裂荚性(PS)是最显著的驯化性状之一,在SNP-GWAS和InDel-GWAS中均监测到相关位点。其中一个PS位点(PS-3.2)在344份材料中存在2个单倍型,Hapll材料的裂荚率更高(48%)(图4b);另外3个PS位点(PS-3.3, PS-4.2, PS-10.3)的不同单倍型之间的裂荚率差异很小。在G和VL的驯化扫描中发现了6个PS相关位点 (图4a),解释了这两个亚种之间PS抗性的差异。基因表达分析发现,大多数裂荚相关候选基因在G型豇豆和V型豇豆荚间的表达模式不同,表明基因的表达可能与PS呈正相关。该发现将为提高豇豆耐裂荚性提供理论基础。
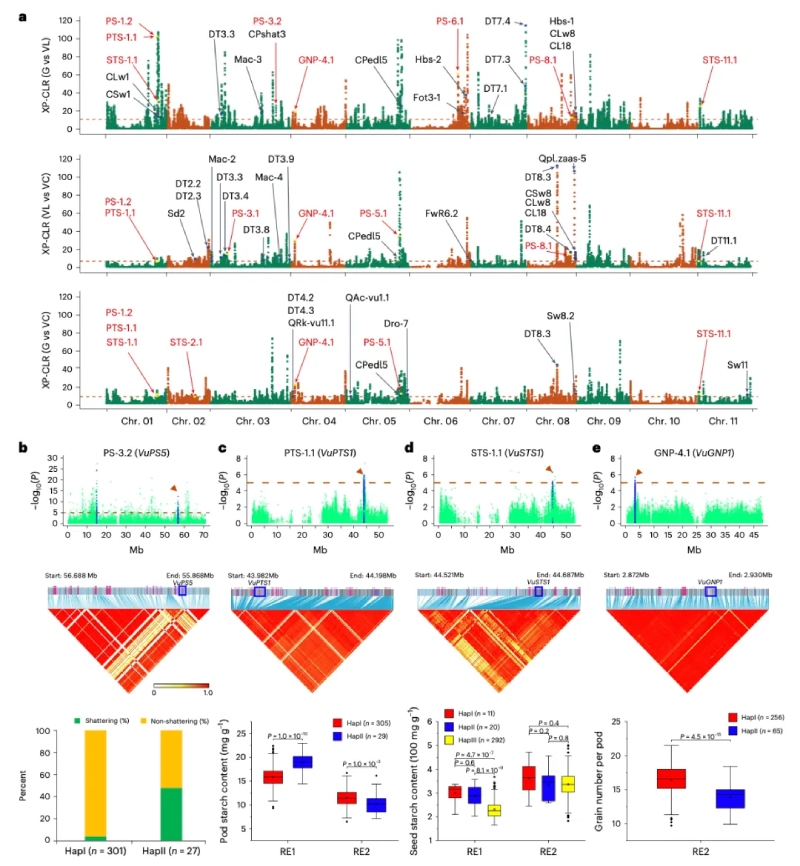
图4-豇豆驯化与改良的选择清除和重要性状GWAS分析
- 豇豆的产量与品质变异
产量相关基因和品质相关基因在3个亚群体(G、VL和VC)中的多态性和分布揭示了豇豆表型分化的遗传基础。荚长(PL)、单荚粒数(GNP)和千粒重(TSW)是影响豇豆产量的重要因素,G亚群的PL通常比VL和VC亚群的PL短得多(图5a)。GWAS分析挖掘到与这3个性状相关的位点和候选基因,并对候选基因单倍型及其表达分析,发现PL差异可能是由于VuPL1-HapII 和 VuPL4-HapI (图5b) 在豇豆驯化和改良过程中强烈选择引起的。在重组自交系(RIL)群体中进一步验证了VuPL1和VuPL4的功能,发现VuPL1似乎比VuPL4在该群体中具有更强的作用。同时基因表达分析发现WAKs基因可能以剂量/转录依赖的方式导致PL差异。粮用豇豆的GNP也通常比菜用豇豆的低,可能是VuGNP2-HapIII这一有利单倍型引起的(图5e)。TSW则可能与VuTSW1和VuTSW2基因的有利单倍型有关(图5f),其功能也分别在RIL群体和F2群体中得到进一步验证。
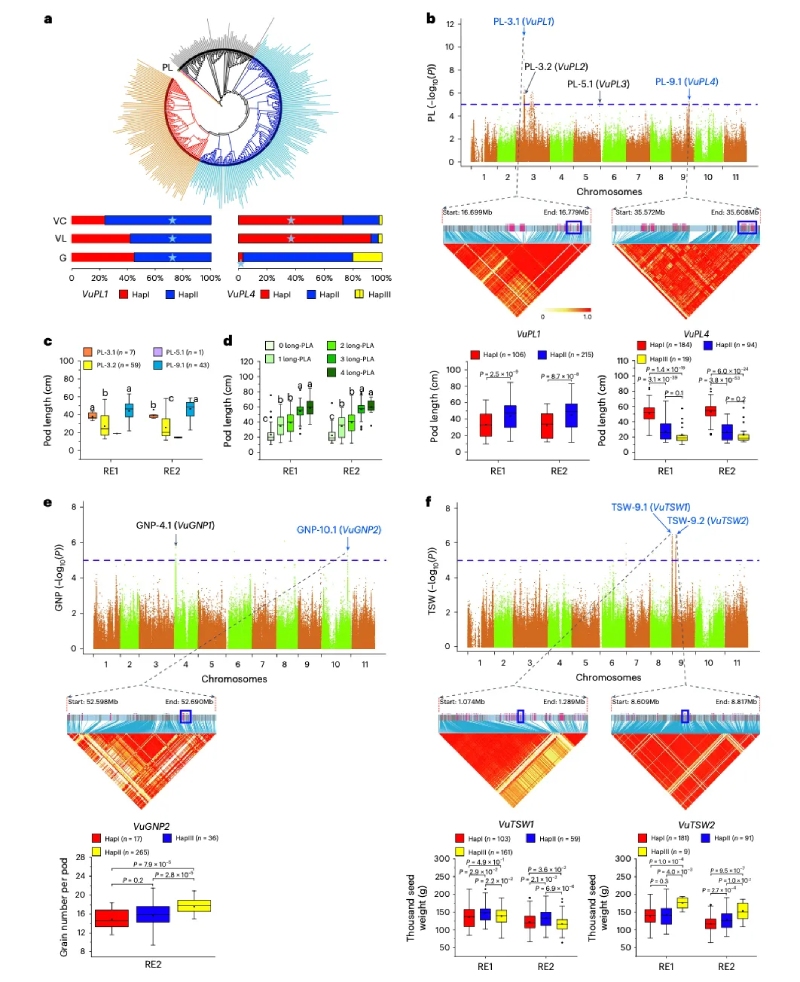
图5-豇豆产量性状相关基因挖掘
可溶性糖、总淀粉和粗蛋白质含量是豆科作物的三个基本品质性状。GWAS分析检测到三个信号与嫩荚可溶性糖含量(PSS)显著相关(图6a),其中VuPSS1可能是功能相关基因,VuPSS2可能通过其顺式调控元件影响PSS含量,VuPSS3-HapIV单倍型是PSS的有利单倍型。嫩荚粗蛋白含量(PCP)差异可能与有利单倍型VuPCP1-HapII和VuPCP2-HapI有关。籽粒可溶性糖(SSS)含量相关的信号(图6e)包含一个FAR1-related SEQUENCE (FAR1)家族蛋白(VuSSS1),FAR1在淀粉合成以及糖的运输和降解中起作用,VuSSS1-HapI为其有利单倍型(图6f)。VuSCP2-HapIII和VuSCP2-HapIV通常导致更高的籽粒粗蛋白含量(SCP),VuSCP1和VuSCP2相似的表达模式提示它们可能影响种子发育中期的SCP表型。籽粒总淀粉含量(STS)GWAS分析检测到5个相关信号(图6i),STS-2.1中的VuSTS2含有一个MYB转录因子,STS-11.1中的VuSTS5编码一个磷脂酰丝氨酸脱羧酶,该酶可能影响植物发育的关键调节因子磷脂酰丝氨酸。两种基因的不同单倍型在两种环境中表现出不同的STSs,这表明它们在很大程度上受环境条件的影响(图6j)。STS-3.1中的VuSTS3编码了一个未知的蛋白,其中VuSTS3-HapIII对STS的影响最大。STS-6.1含有一个NAD依赖性蛋白去乙酰化酶VuSTS4,可能影响淀粉的生物合成和调控,而VuSTS4-HapI对STS的作用更强。以上结果有潜力成为粮用豇豆和菜用豇豆产量和品质改善的遗传资源。
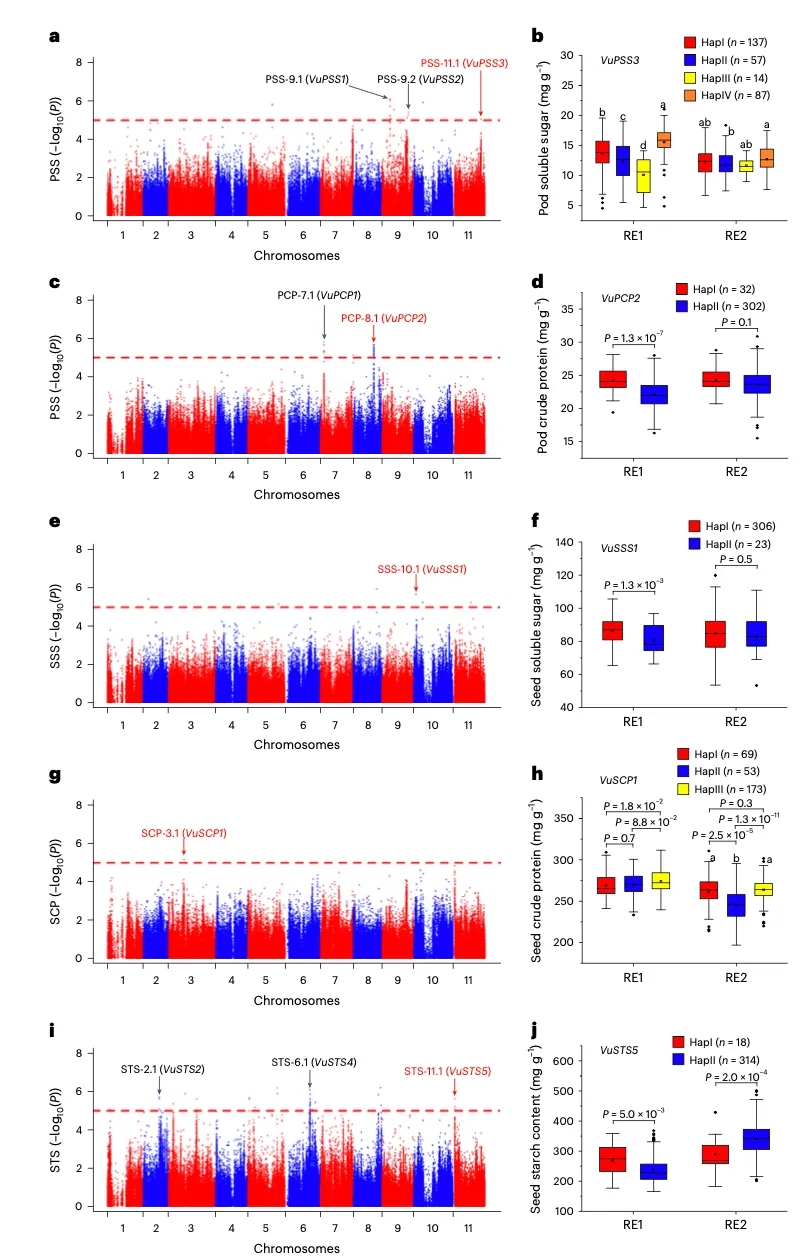
图6-豇豆品质性状相关基因挖掘
研究总结
本研究结合PacBio、Hi-C和二代测序,组装了粮用豇豆和菜用豇豆的染色体水平基因组。对包括地方品种、野生品种和育成品种的344个材料进行二代测序,以阐明豇豆基因组的系统进化。此外,还进行了全基因组关联分析(GWAS),确定关键产量和品质性状相关基因。该研究揭示了两个亚种之间基因组结构变异(SVs)的全图谱,为豇豆在全基因组选择下的驯化与改良提供了见解。产量性状和品质性状的差异基因组选择将有助于建立粮用豇豆和菜用豇豆双向改良的遗传资源。
参考文献:Wu, X., Hu, Z., Zhang, Y. et al. Differential selection of yield and quality traits has shaped genomic signatures of cowpea domestication and improvement. Nat Genet (2024).



 京公网安备 11011302003368号
京公网安备 11011302003368号