

种质资源是自然遗传多样性的重要来源,丰富的动植物种质资源为遗传资源和育种研究提供了基础,但同时也给遗传资源的保存、研究和利用带来了困难。为此,Frankel等人于1984年提出了核心种质(core collection)的概念。构建核心种质资源库是有效探索和保护遗传资源新变异的重要途径,近几十年来,已经在多种植物上建立了核心种质资源库,但是传统的方法主要是通过形态学性状和地理来源来判断,这种方法存在明显缺陷。随着高通量测序技术的不断发展,更有效的方法是基于基因型鉴定来筛选核心种质,利用高分辨率的分子标记类型,可以提高遗传材料内及材料间遗传相似度和杂合度的鉴别能力,更加有利于核心种质的构建。
一、核心种质资源库概念:什么是核心种质?
核心种质资源(core collection),即保存的种质资源的一个核心子集,是采用一定方法,从保存的某一物种种质资源中抽取的一个核心子集,以最少的遗传资源样本量最大限度地代表包括地理分布在内的整个资源群体的遗传多样性,而未列入核心种质的其它资源材料则作为保留样本予以保存。因此核心种质可以作为种质资源群体研究和利用的切入点,从而提高整个种质库的管理和利用水平。
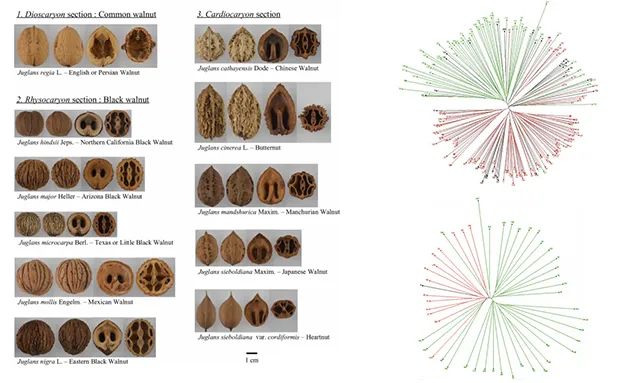
胡桃核心种质筛选(Bernard et al., 2018)
二、核心种质研究进展
1984年,Frankel等人首次提出了核心种质(core collection)的概念。近几十年来,核心种质发展迅速,已在多种植物上建立核心种质,特别是近几年分子标记和测序技术的广泛应用使得基于高密度SNP标记构建核心种质成为可能,对种质资源的群体结构和遗传多样性研究也愈加深入。如(Girma et al., 2020)等用879,407个SNP对2010份埃塞俄比亚高粱种质资源进行核心种质资源鉴定,选择了387个(种质资源的20%)种质作为核心种质;(Kumar et al., 2020)等用3,565,117个高质量SNP对3004份水稻进行核心种质鉴定,得到一个包含520个种质的微核心种质,结果表明其代表了原有样品的所有表型和地理来源;(Milner et al., 2019)等对22,621份库存大麦种质资源基因组多样性进行研究,共选出1000份作为其核心种质集。除此之外,还有芝麻、小麦、棉花、甘薯、葡萄、杏、黄瓜、茄子、杉木、马尾松等多种植物成功构建了核心种质资源库。
三、核心种质研究技术路线
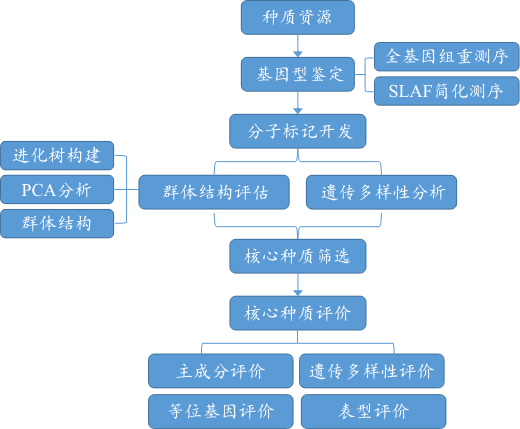
核心种质研究技术路线
四、核心种质材料选择
种质资源分为两个层次,一是某个物种的所有种质资源;另一是特定种质资源。对于一个物种所有的种质资源,应尽可能选择整个较多的该物种的材料;对于特定的种质资源,可以选取不同地理位置性状、表型性状变异广泛的材料、品种之间具有差异(野生种/驯化种/地方品种等)的材料。材料的选择直接决定了核心种质的表型和遗传变异组成,因此,选择用于筛选核心种质的材料应尽可能地保证种质资源的遗传多样性。
五、核心种质构建方法
如前所述,核心种质是要以最少的遗传资源数量最大限度地代表包括地理分布在内的整个资源群体的遗传多样性。所谓核心种质构建是指采用一定的方法从现有的种质资源的总样品中提取符合上述目标的核心种质。最初,包含形态和农艺性状的表型数据被用来创建核心集合,而现在分子标记作为测量遗传变异的中性工具已成为选择的工具。核心种质的构建有不同的方法,(Lee et al., 2020)利用PowerCore,根据从南瓜基因组中均匀分布的2071个SNP计算出的遗传变异,从595个原始种质中选出67个核心种质;(Xu et al., 2020)利用 Core Hunter II的Mstrat策略,从204个青藏高原青稞种质中筛选得到41个核心种质;(Liu et al., 2020)利用LDSS方法对湖北省内分布份208个重齿当归进行核心种质鉴定,发现包含42个种质的核心种质集能更好地代表原始种质。从以上可以看出,不同的研究者在在不同物种核心种质构建过程中所采用的方法不尽一致,不管用哪种方法,前提都需要对整个种质资源的群体结构和多样性组成有足够的了解,根据遗传变异标记(SNP)数据,结合多种评估措施(Modified Rogers distance、Shannons Diversity Index等)进行加权处理,筛选出具有高多样性、高代表性和高等位基因丰富度的材料。
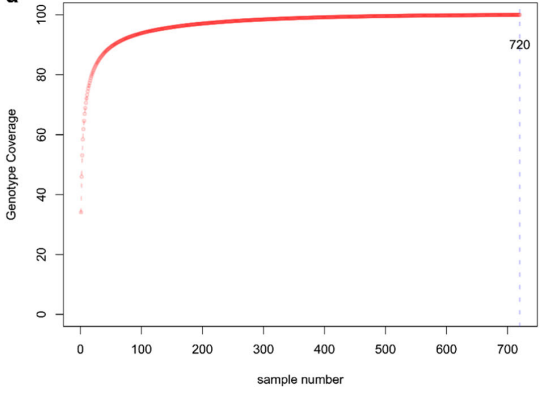
1234份黄瓜核心种质鉴定(Wang et al., 2018)
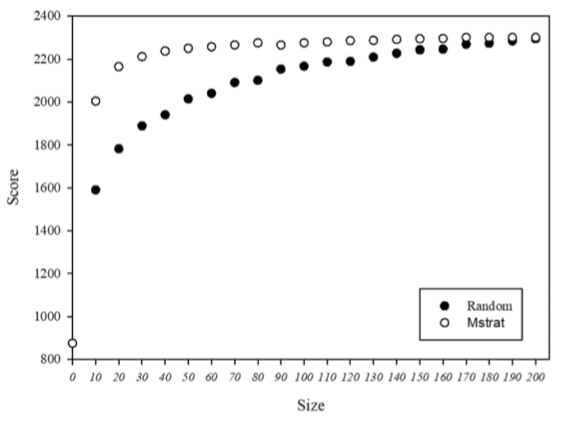
204份青藏高原青稞核心种质鉴定 (Xu et al., 2020)
六、核心种质的评估
构建好的核心种质采用何种方法去评估核心样品对整个种质资源多样性的代表性同样是核心种质研究中的重点。目前,大多数研究者结合不同的方法对核心种质进行评估验证,通过对原始种质材料和筛选的核心种质材料进行主成分分析,评估种质筛选的准确性,原则上,基于核心种质绘制的主成分图和所有材料的分布图趋势吻合,就能说明筛选结果的合理性;计算常规的遗传多样性指标:观测杂合度(Observed heterozygosity)、期望杂合度(Expected heterozygosity)、Nei遗传多样性(Nei diversity index)、香浓维纳多样性指数(Shanon-Wiener index)、多态性信息含量(PIC),对原始种质材料以及筛选的核心种质的遗传多样性进行评价;另外还包括等位基因评价和表型评价。
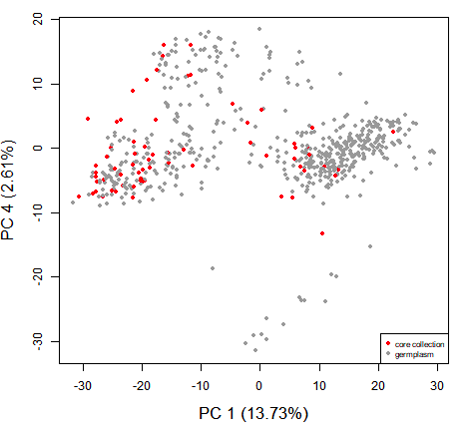
南瓜核心种质PCA分析评估(Lee et al., 2020)
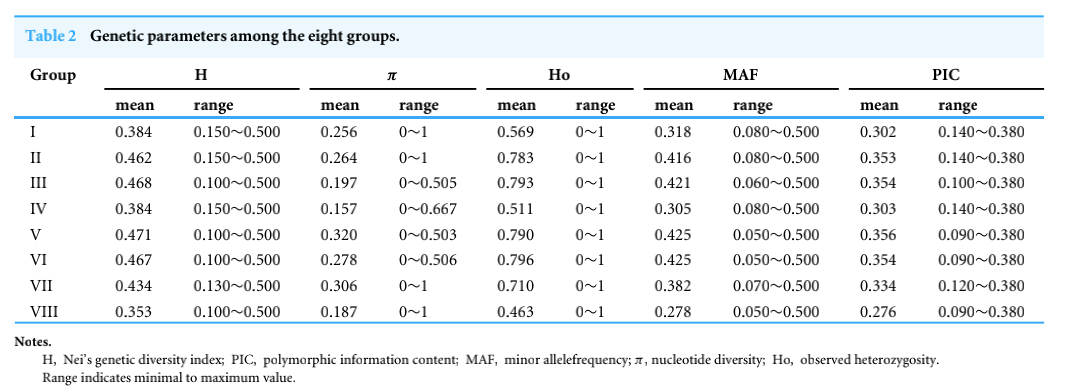
芋头核心种质遗传多样性评价估(Wang et al., 2020)
七、总结
核心种质可以最大限度地去除原始种质资源中的重复,以最少的种质材料代表原始种质资源的全部或大多数遗传多样性和地理来源,为当前越来越多的种质资源收集、评价和利用带来了便利,特别是随着基因组学的发展,测序成本的极速下降,使得大规模群体测序得以实现。利用高分辨率的分子标记,提升遗传材料间遗传相似度和杂合度的鉴别能力,从而选择能够代表整个种质资源基因多样性的大小合理的核心种质材料,更加有助于种质资源的保存、管理、使用。使得我们可以有重点地进行优异种质的研究,结合GWAS分析、遗传进化分析、QTL定位、特有标记开发等方法进一步进行基因的挖掘与克隆,提高种质资源的利用效率。
八、案例分享
Genome-wide assessment of population structure and genetic diversity and development of a core germplasm set for sweet potato based on specific length amplified fragment (SLAF) sequencing [1]
研究材料:197份甘薯种质资源(50个地方品种和147个栽培品种)
主要内容:本研究利用SLAF-seq技术对甘薯的50个地方品种和147个栽培品种进行简化基因组测序,开发共得到了62,363个SNP标记,基于这些SNP对本研究的197个甘薯种质资源进行群体结构和遗传多样性评估,群体结构分析将材料分为三组;利用系统发育树评估材料之间的遗传关系,同样将所有材料分为三个组。主成分分析 (PCA) 表明,这些种质根据其种群结构进行分布。此外,计算了种质间的平均遗传距离、平均多态信息含量 (PIC) 和平均次要等位基因频率 (MAF) 。使用 CoreHunter 软件,鉴定得到了包含39 个材料的核心种质,约占总种质资源的19.8%。并对核心种质从PCA分析、遗传多样性、等位基因数层面进行了评价。本研究开发的甘薯核心种质将为未来甘薯育种改良提供宝贵的种质资源。
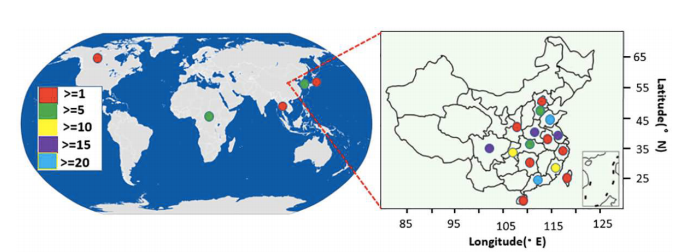
197份甘薯地理来源
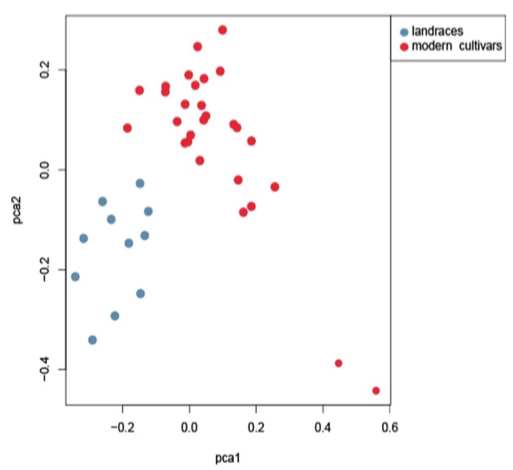
马尾松核心种质
马尾松GWAS分析
Core set construction and association analysis of Pinus massoniana from Guangdong province in southern China using SLAF-seq [2]
研究材料:149份马尾松种质资源(来源于广东省9个不同的地点)
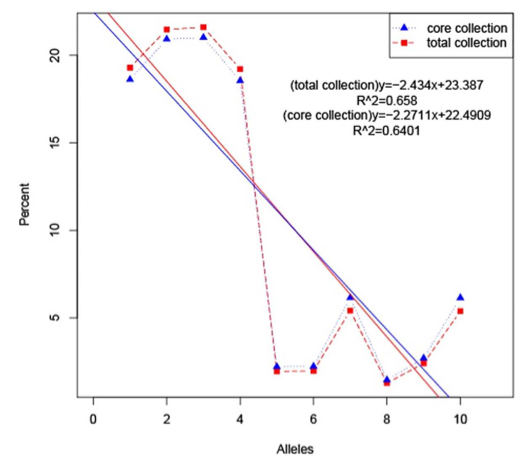
主要内容:在本研究中,采用SLAF-seq技术对从中国广东收集的149份马尾松(Pinus massoniana)材料进行测序,从599,164个多态性SLAF标签中鉴定了471,660个SNP标记。群体结构分析表明,149份马尾松不能划分成明显的亚种群。使用遗传距离和种群结构来选择核心种质,筛选出包含29个材料的核心种质,分别与树脂产量和木材体积相关。对122份马尾松材料进行关联分析,包括不同高度(HT)、胸径(DBH)、树脂质量(RW)、木材体积(VW)和树脂产率(RYC),使用mrMLM、FASTmrMLM、FASTmrEMMA和ISIS EM-BLASSO检测到大量的SNP与性状HT、DBH、RW和RYC显著相关。马尾松核心种质为未来的改良育种提供宝贵的资源。
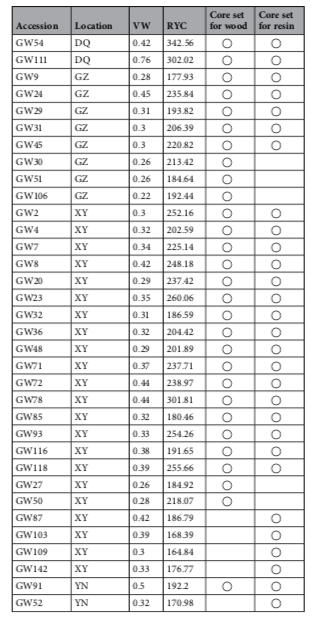
马尾松核心种质
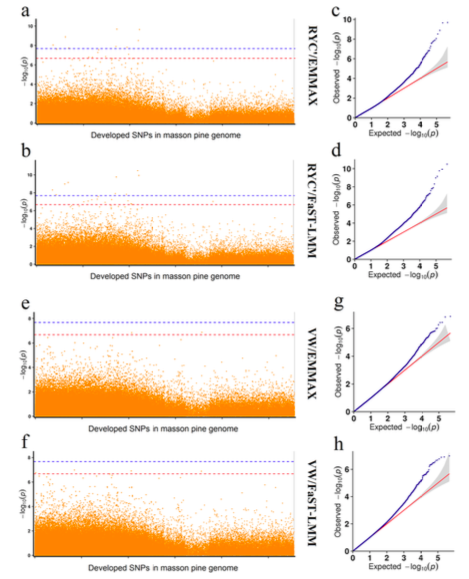
马尾松GWAS分析
如果您对种质资源构建项目感兴趣,欢迎点击下方按钮联系我们

相关阅读
参考文献
[1] Su W, Wang L, Lei J, et al. Genome-wide assessment of population structure and genetic diversity and development of a core germplasm set for sweet potato based on specific length amplified fragment (SLAF) sequencing. PLoS One. 2017;12
(2):e0172066.[2] Bai Q, Cai Y, He B, Liu W, Pan Q, Zhang Q. Core set construction and association analysis of Pinus massoniana from Guangdong province in southern China using SLAF-seq. Sci Rep. 2019;9(1):13157.



 京公网安备 11011302003368号
京公网安备 11011302003368号