

基因组作为生命信息的承载体,蕴含着每种生物的全部遗传信息。近年来,随着测序技术的不断发展,基因组学研究已经逐渐成为一项非常重要的基础研究,而自然界中每个物种的基因组都有望被成功破译。为了能让更多想要进行基因组项目的科研工作者都能有个初步了解,今天小编整理了一些常见问题,一起来看看吧!
Question01:为什么要构建基因组?
Answer:基因组表示的是一个物种内全部的遗传信息,没有参考基因组使得关键基因无法被挖掘,调控机理难以被解析,成为科研的掣肘。而早期构建的参考基因组质量往往较差,导致①组装不完整,可能遗失相当多的基因片段,想要的基因因为未被组装到而被错失。②连续性较差,短片段较多,且不利于研究由较长片段形成的与功能相关的基因。③拼接准确性有偏差,较短的片段在拼接时易因序列重复导致排序错误,从而影响后续相关研究的顺利进行。甚者,所研究品种与已发表参考不同使得研究受到阻碍①相同的种下不同的品种/品系/变种比对率低,可用数据少;②雌雄性别差异,公布只有单个性别,找不到性别相关区域。
Question02:基因组的组装难易程度主要由哪些方面影响?
Answer:①基因组大小。基因组越大,对应的重复序列往往越丰富,导致拼接的难度越高;②杂合度与重复序列比例。相同大小的基因组下,杂合度和重复比例越高,基因组组装的连续性和完整性会越低(高杂合的基因组往往无法合并姊妹染色体,导致组装的结果偏大,杂合位点容易拼接断裂使得连续性降低,而重复序列在组装中会被折叠,使组装中出现缺口、错误,导致组装的结果偏小)。因此通常会需要适当增加测序深度以覆盖这些复杂的区域。③基因组的倍性和倍型。难易程度由易至难分别为:二倍体>异源多倍体>同源多倍体。
Question03:如何知道物种基因组大小?
Answer:①已发表过基因组的可通过NCBI网站查询:https://www.ncbi.nlm.nih.gov/②未发表基因组的通过流式网站查询:植物–https://cvalues.science.kew.org/ ;动物:http://www.genomesize.com/③进行流式、survey(调研图)进行分析
Question04:Survey是什么?可否不进行?
Answer:①Survey以二代测序技术为基础,基于小片段文库进行低深度测序,通过K-mer分析,快速获得基因组大小、杂合度、重复序列比例等基本信息的研究方法。为制定该物种的全基因组de novo测序策略提供有效依据。②survey的二代数据具有纠错和评估的重要作用,除非是已知基因组大小的单倍体等背景较为清晰的物种情况下,通过HiFi模式组装,并且不需要做二代数据评估,可以考虑不进行,否则建议为必须进行。
Question05:为什么三代测序样品要与二代survey测序样品来自同一个个体?
Answer:①不同个体间会存在一定差异,若选材差异大可能会影响到三代测序策略的制定②二代数据需为Nanopore/Pacbio CLR模式基因组进行纠错,避免因个体间序列差异影响纠错效果③二代数据需回比组装完成的基因组来评估该基因组组装的完整性,避免因个体间序列差异降低比对率。
Question06:是否必须等Survey分析完之后才能启动三代测序?
Answer:针对已知倍型倍性、已明确基因组大小(通过流式等方式)或已经发表过同品种、近缘种材料的项目,可以同时启动survey与三代测序,节约时间成本,使项目更快的推进。
若物种背景尚不完全明晰,需要先完成survey,再开展三代测序组装。基于该物种基因组的大小、杂合及重复序列比例来制定合适的三代测序深度与数据量。
Question07:什么是HiFi测序?跟以前传统的测序模式有什么区别?
Answer:目前PacBio Sequel II平台可提供CLR library和HiFi library两种模式,CLR文库是传统的组装时构建的文库类型,采用20 Kb、30 Kb等长片段类型的DNA进行文库构建,获得Subreads,单碱基准确率在85%左右,因此在基因组组装前,需要通过canu等软件进行数据纠错后用于下一级应用。HiFi reads(High fidelity reads)是兼顾长读长和高准确度的测序序列,即15Kb-20Kb片段文库,进行单一片段多轮测序的方式来提升准确性,单碱基准确率可达99%。HiFi测序结合Hifiasm及HiCanu等软件,可在较少的资源消耗下快速完成基因组的组装,并保证结果的高准确性和连续性,尤其是对超大、超高杂合等复杂基因组具有明显优势,同时也为高精度基因组注释、变异检测等应用提供了更有利的支持。
Question08:为什么PacBio HiFi测序只用几十乘深度即可满足分析需求
Question09:什么是染色体版本基因组?如何构建染色体水平的基因组?
Answer:染色体版基因组是指:将三代测序等方式拼接到的基因组序列分配至染色体组中,明确位置与方向向,使组装的基因组达到染色体水平。常用的方式主要为Hi-C、遗传图谱或光学图谱。
Question10:为何使用Hi-C进行染色体的挂载?其原理是什么
Answer:Hi-C技术将线性距离远、空间结构近的DNA片段进行交联富集后Pair-end测序,根据同一条染色体上的染色质片段互作频率更高,不同染色体间的互作频率较低的特点,推导出基因组的三维空间结构和基因之间可能的调控关系。利用Hi-C测序数据将Draft genome序列进行染色体群组的划分,并确定各序列在染色体上的顺序和方向,使基因组组装组装水平提升到染色体水平。
与其他技术相比,Hi-C具有以下优点,因此具有更广阔的发展①无需群体,单个个体就能实现染色体挂载;②标记密度更大,锚定染色体效率高,挂载率≥90%;③可以对已组装的基因组进行纠错;④分析周期短,准确性高
Question11:哪些参数可以评估构建的基因组?
Answer:①基因组大小及连续性(N50):基因组组装大小与调研图一致、N50值越高越好。(通常contigN50值≥1Mb即可满足绝大多数分析需求)②二代回比率:将二代高通量测序得到的短序列与组装得到的基因组比对,通过统计比对率,可评估组装基因组的完整性。③Busco/Cegma等数据库评估:在组装得到的基因组上查找软件数据库中的保守基因,通过找到的保守基因比例,评估基因组上基因组装的完整性。④LAI评估,鉴定完整LTR-RTs占比。
Question12:为什么做基因组需要测RNA?还需要提供混组织的样品?
Answer:在基因组组装完之后需要对基因组的结构与功能进行预测与注释。目前基因预测是从头预测(基于结构)、同源预测(基于近缘物种)、转录组(基于表达基因)三部分结合进行研究,转录组所表达的即为最真实的情况,因此在基因预测的过程中具有重要的作用。而由于转录表达的时空特异性与组织特异性,为了获得更全面的信息是需要进行多个组织部位混合检测。
Question13:基因组做完之后可以开展什么研究?
Answer:基因组完成后可以进行比较基因组学分析,与近缘物种进行宏观进化研究,其内容主要包括:(1) 基因家族聚类,分析特有、共有基因和基因家族;(2) 基因家族扩张收缩分析;(3) 系统发育树的构建;(4) 物种分化时间推算;(5)LTR形成时间估算(一般为植物基因组的分析项);(6)全基因组复制事件(一般为植物基因组的分析项);(7)选择压力分析;(8)共线性分析。具体可见涨知识啦!比较基因组学研究那些事
尾声
百迈客自2009年成立以来深耕于群体遗传研究,同时具有近10年基因组分析经验。现已在基因组、Hi-C、遗传图谱等技术上拥有实验+生信分析双保障。并与国内外70余所科研单位在国际期刊合作发表500余篇高质量文章,累计影响因子3000+。若您也对基因学研究感兴趣,或是还有其他问题想进一步的了解,可以联系当地销售经理或致电400-600-3186
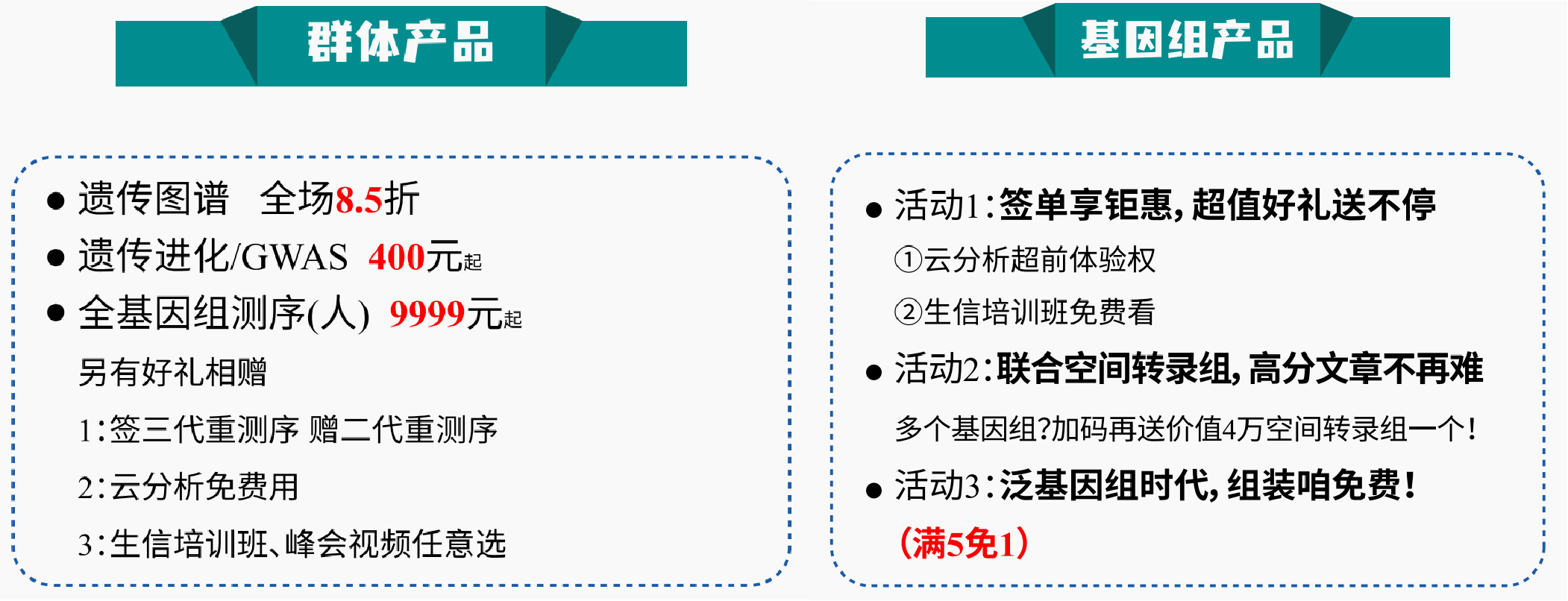
 百迈客特在年末之际,推出多项优惠活动,助力科研!
百迈客特在年末之际,推出多项优惠活动,助力科研!




 京公网安备 11011302003368号
京公网安备 11011302003368号