

中文名: 通过全长转录组对斑马鱼转录组进行高分辨率注释
英文名: High resolution annotation of zebrafish transcriptomeusing long-read sequencing
杂志:Genome Research 2018 09
影响因子:10.101
研究背景
斑马鱼是一种重要的模式生物,已被广泛用于胚胎发育等多方面研究。斑马鱼 发育的第一阶段完全由母系提供的 mRNA 和蛋白质指导,随着胚胎发育的进行,发育由母 源因子控制转为由合子基因产物控制。这一过程中,母源因子逐渐清除,合子基因组激活 (ZGA)并开始转录。目前通过转录组 RNA-seq 技术对早期胚胎发生过程取得了大量的研究 成果,但是由于短读长 RNA-seq 技术的局限性,对斑马鱼转录组的相关基因注释和可变剪切 等的研究有很大的不足。而目前出现的长读长的三代全长转录组可以通过直接读取全长转录 本的序列克服这些不足,以揭示早期斑马鱼转录组的其他新颖性和复杂性,从一个新的角度 对斑马鱼转录组进行大量扩展研究和验证。
材料方法
1、实验材料:选择斑马鱼受精卵,通过形态学标准进行分期,鉴定胚胎前期(256 细 胞期)和后 ZGA 期(6hpf)。通过注射 0.2nmol 的 RNA 聚合酶抑制剂α-鹅膏毒素来处理 1 至 4 个细胞的胚胎获得处理组。
2、转录组测序:未处理组的胚胎选取胚胎前期和后 ZGA 期胚胎进行转录组测序,每个 时期 3 个生物学重复。
3、全长转录组测序:选取正常的胚胎和 RNA 聚合酶抑制剂α-鹅膏毒素处理的胚胎进 行三代全长转录组测序。
研究结果
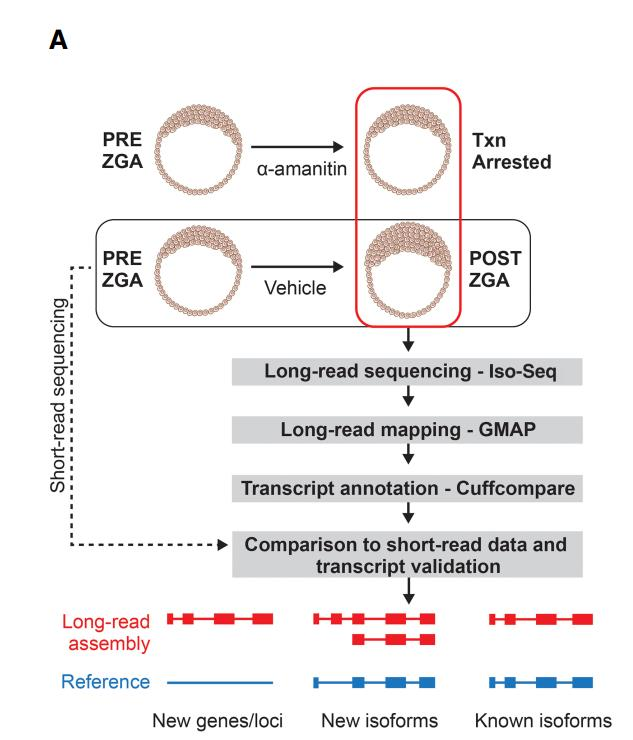
1、试验系统的设计与全长转录组数据概况
将杂交得到的斑马鱼胚胎通过形态学标准进行分期,将经过α-amanitin 处理以及未经处 理的胚胎分别转录组和全长转录组测序。使用 Iso-Seq pipeline 将得到的长片段的全长转录 组数据进行聚类和组装得到全长转录本。为评估全长转录组数据将得到的转录本数据库 通过 GMAP 与斑马鱼参考基因组(GRCz10)数据进行比对,发现 18,777 个转录本成功比对,仅有 3.6%的数据无法比对,而转录组数据中至少 20%的数据无法比对。
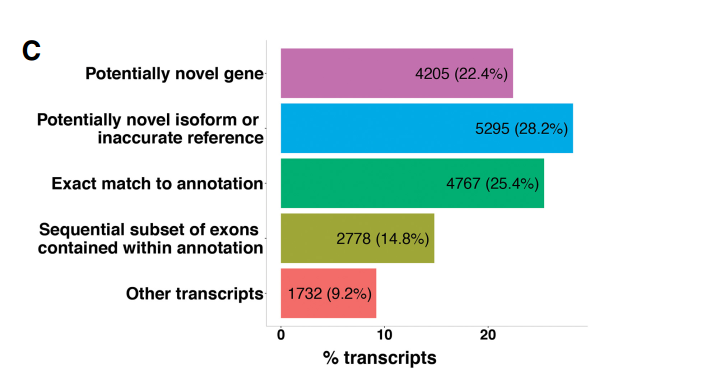
将全长转录组得到的转录本通过 Cuffcompare 软件进行注释分析,发现 4767 个转录本 (25.4%)与注释完全匹配;9500 个转录组(50.6%),可能代表新的转录本,其中 4205 个 转录本(22.4%)是新转录本(NTR),与参考注释没有重叠;5295 个转录本(28.2%)为先 前注释基因的未发现转录本;2778 个转录本(14.8%)为连续注释外显子集合;1732 个转 录物(9.2%)被认为来源于难以产生转录本的区域,暂时不予考虑。
2、新型转录区域的鉴定
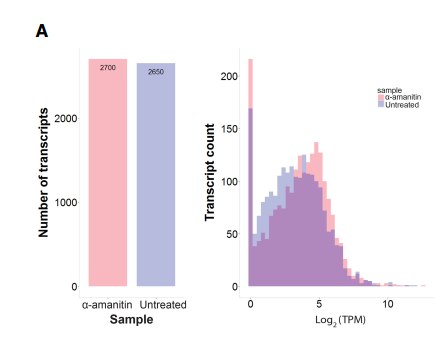
为了对新发现的转录本进行分析,文章将短读长转录组中的数据比对到加入了全长转 录组的参考基因组数据库,发现在未经处理和α-amanitin 处理的样品中通过全长转录组发 现的超过 2000 个主要不重叠的新转录本中分别有 89%和 86%有转录组数据支持。并且发 现,在高表达的基因中,短读长转录组数据的外显子比对率从从 68%提高到了到 85%。通 过 CPAT 对新发现转录本进行编码潜能预测,在 4205 个新型转录本中,3255 个序列具有高 编码潜力的,使用 Pfam 的验证结果也基本类似。利用这些数据我们鉴定到了一个 H2AFX 新 的转录本仅在处理后的样本中出现,HIST2H2BE 基因的新鉴定到的转录本仅在未处理的赝本中出现。
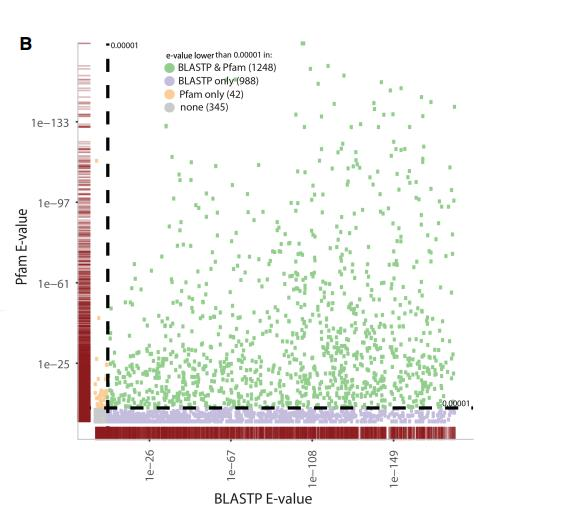
3.新转录本编码潜能预测
通过 CPAT 对新发现转录本进行编码潜能预测,在 4205 个新型转录本中,3255 个序 列具有高编码潜力的,使用 Pfam 的验证结果也基本类似。利用这些数据我们鉴定到了一个 H2AFX 新的转录本仅在处理后的样本中出现,HIST2H2BE 基因的新鉴定到的转录本仅在未处 理的赝本中出现。
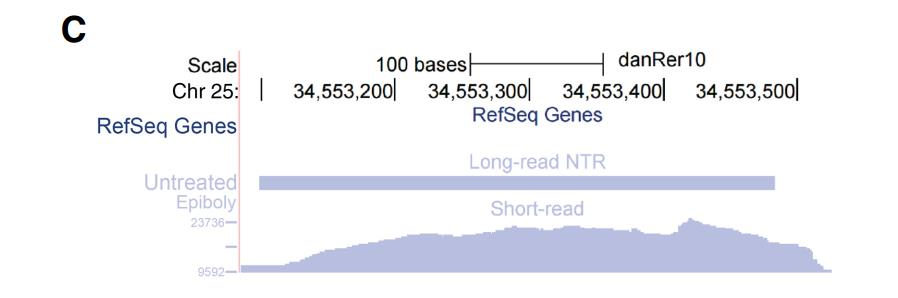
对于非蛋白质编码的 NTR,通过 Rfam 数据库进行鉴定和注释,鉴定了 76 个与 Rfam 数 据库相匹配的转录本,发现了一个与 Rfam 数据库中的 mir-548 相匹配的转录本,该转录本 在α-amanitin 处理的样本中特异表达,推测它主要存在于母源 RNA 中。
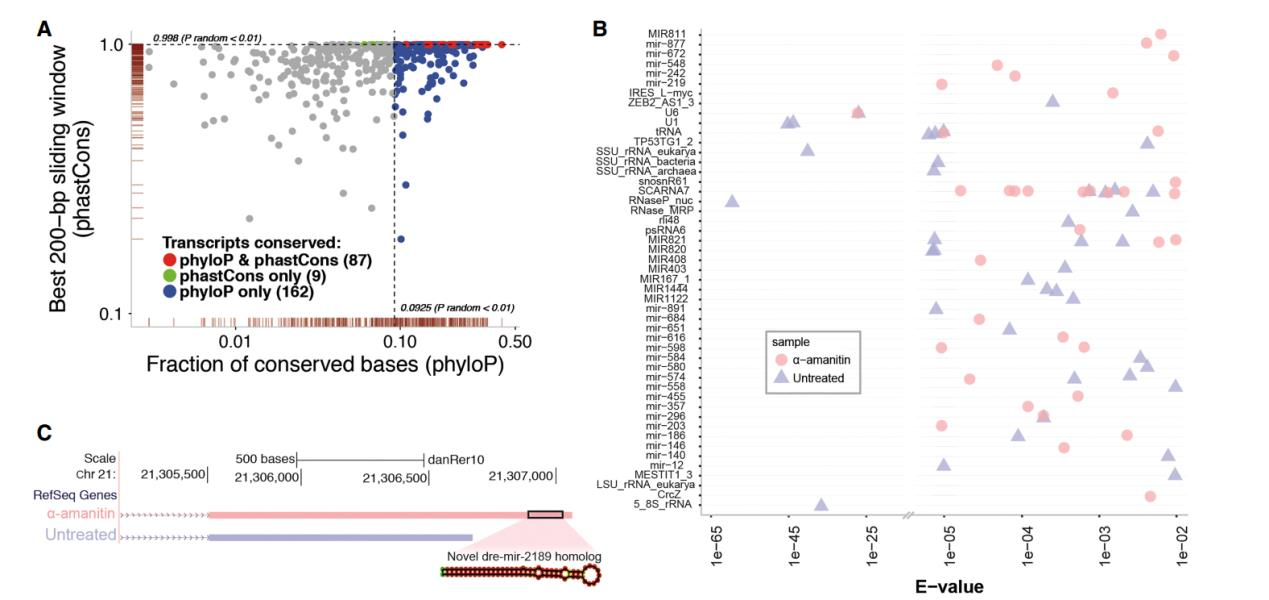
在所有新鉴定到转录本中,通过合格的筛选和注释标准,总共鉴定到了 2278 个新的具 有蛋白质编码能力的转录本,261 种新的非编码的转录本,些新注释的序列将有助于研究它 们在 ZGA 和早期发育中的作用。
4.已注释基因的新转录本分析
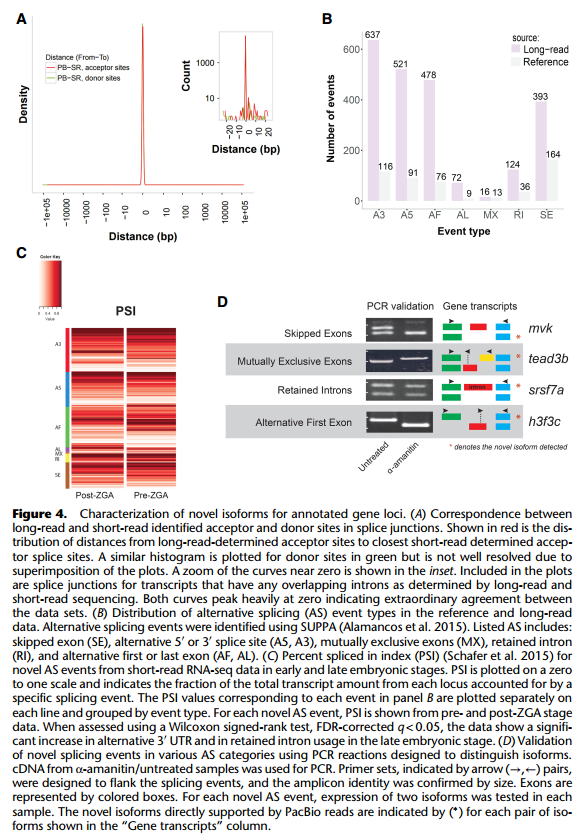
作者接着对全长转录组对已注释基因鉴定到的未记录的转录本的可变剪切形式的研究。 由于斑马鱼参考基因组对可变剪切的注释较少,文章使用短读长的转录组数据对可变剪切位 点进行验证。作者使用 STAR 软件分析转录组数据,预测了 3000 个可变剪切位点,其中 99% 可以与全长转录本数据吻合。剪接位点的这种近乎完美的一致性支持证明了全长转录组鉴定 的外显子 – 内含子边界的准确性,并强烈支持新检测到的可变剪接形式的有效性。通过全 长转录组共发现了 2000 种新型可变剪切类型,其中已注释基因的新型可变剪切共 1835 中, 平均每个基因的可变剪切类型提升 50%。
5.新转录本的功能鉴定
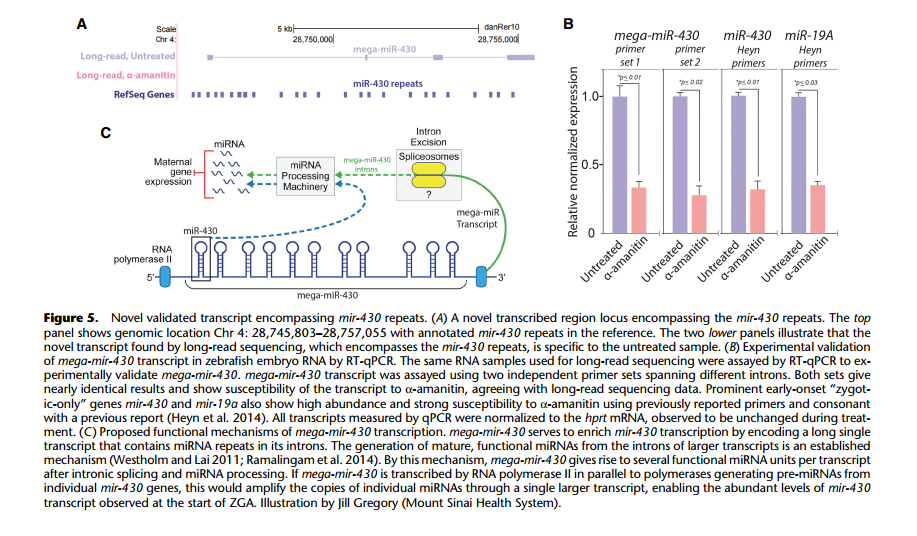
通过全长转录组鉴定到的大量新转录本中包括了一种重要 miRNA- miR-430,mir-430 可能是第一个表达的合子基因,它通过对所有母源转录基因进行的特异性沉默和降解将合子 基因的启动和母体程序的消除连接起来。在以前的研究中,对其功能研究较多,而对于 miR-430 的来源研究较少。作者鉴定到了一个新的跨越 9kb 基因组序列的四外显子转录本, 该转录本重叠 22 个 mir-430 重复序列,被称为“mega-mir-430”。 在α-amanitin 处理中没 有检测到 mega-mir-430,并通过 qPCR 进行了验证,推断它来源于合子基因组。从每个 mega-mir-430 转录本产生多个 mir-430 的拷贝,这个发现解释了合子基因激活时 mir-430 高表达的原因。
小结
作者通过全长转录组对斑马鱼的转录信息进行了更加全面和深入的分析,利用三代全 长转录组和二代转录组大大提高了斑马鱼转录组注释的分辨率,同时为分析基因复杂的可变 剪切形式提供了有力的研究工具,证明了全长转录组对于斑马鱼合子基因激活的研究的具有 重要推动作用。




 京公网安备 11011302003368号
京公网安备 11011302003368号