

微生物组学的研究从最初的单个微生物形态研究,逐渐深入到微生物的DNA、RNA、蛋白、代谢层面,而这些也衍生出来了一系列的组学研究技术。从初始的形态学观测,到DNA层面的扩增子,宏基因组技术研究其微生物组成和功能,进而到宏转录组、宏蛋白组、宏代谢组等宏表型组学的研究。多样性和宏基因组指明【who is there】有哪些微生物和【what are they doing】这些微生物有哪些功能?而蛋白代谢层面,则可以解决【what have really happened】的问题,哪些过程是实际产生的,这些与表型又有着什么样的关联。
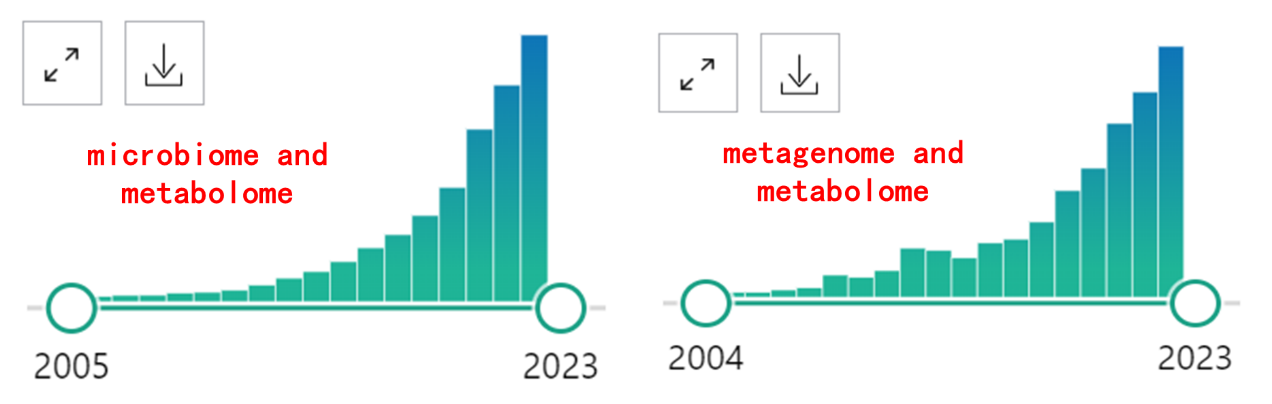
而近年来,关键词“microbiome and metabolome”、“metagenome and metabolome”也是逐渐备受关注的热点话题,PubMed搜索发文量也是逐年上升的趋势。
那么如何来进行微生物和代谢组联合分析呢?联合分析的图表又怎么解读?莫慌,本次小编就为大家带来详细介绍。
首先,我们进入百迈客云平台,打开代谢组分析结题报告,在报告左侧找到联合分析模块,然后选择与微生物联合分析个性化,再在右侧选择需要参与分析的微生物报告,之后进入第二步参数设置,示例图如下:
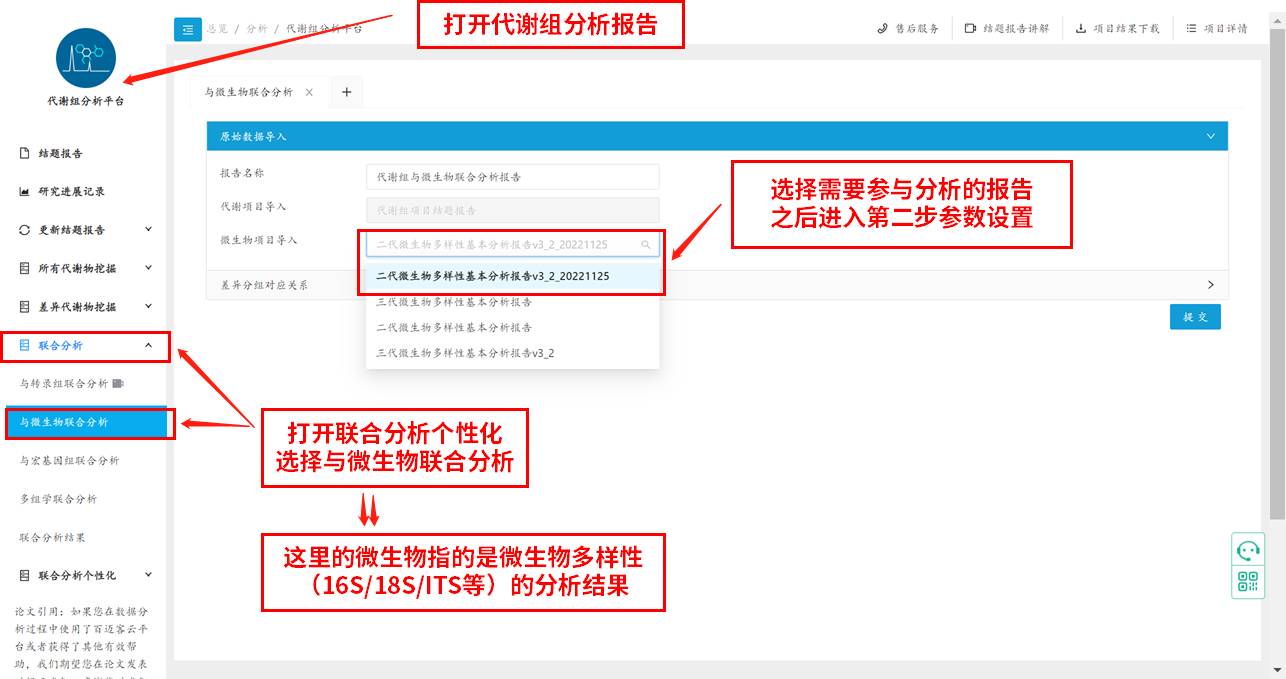
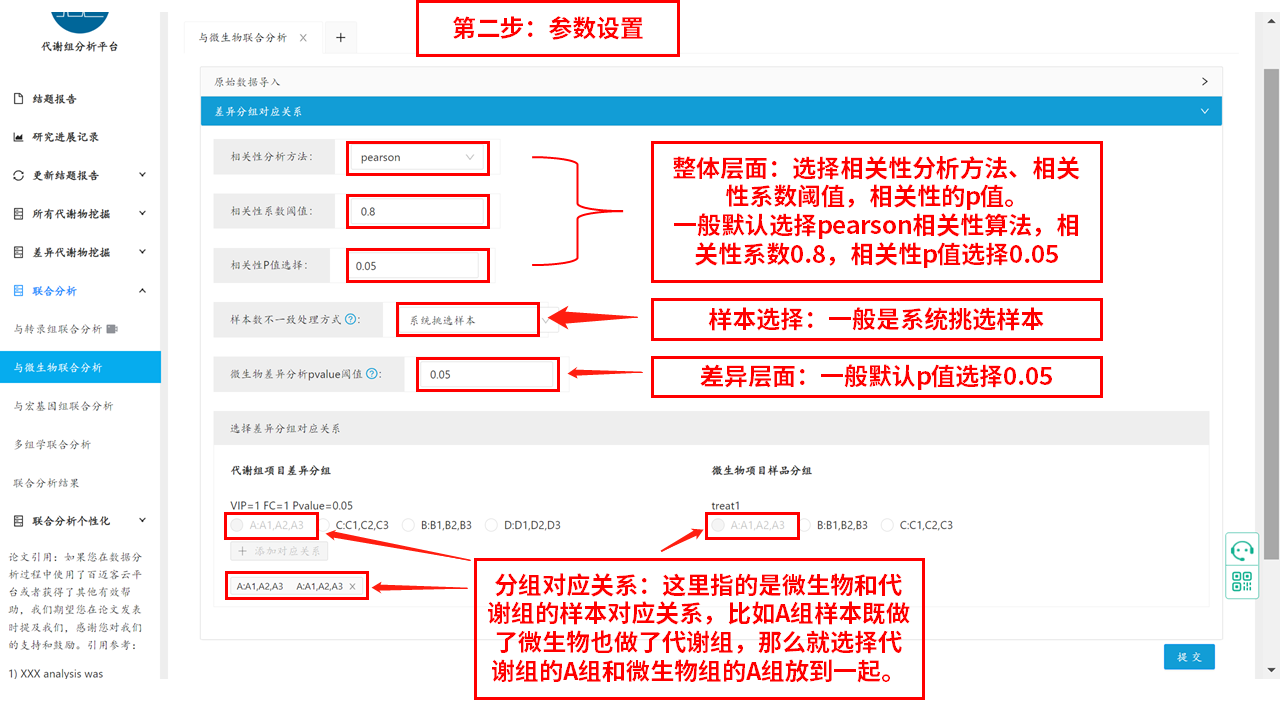
第二步参数设置:整体层面选择相关性分析方法、相关性系数阈值,相关性的p值。一般默认选择pearson相关性算法,相关性系数0.8,相关性p值选择0.05。分组对应关系:这里指的是微生物和代谢组的样本对应关系,比如A组样本既做了微生物也做了代谢组,那么就选择代谢组的A组和微生物组的A组放到一起,具体示例图如下:
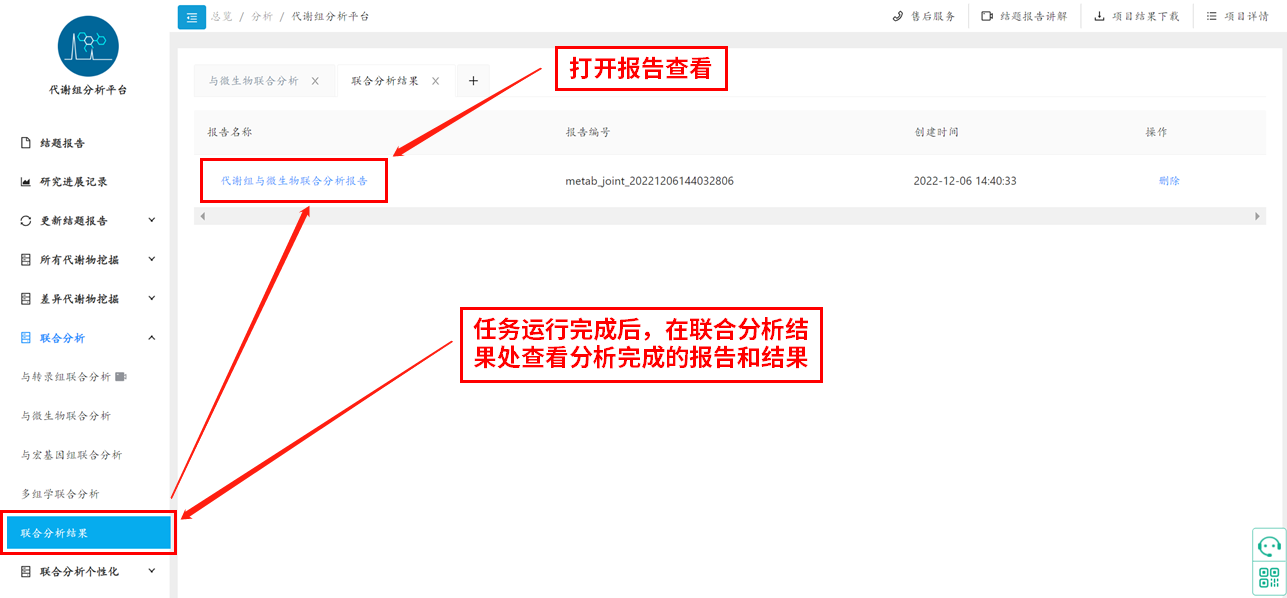
提交分析:等待分析完成获得分析报告结果,一般任务运行周期1-2h。任务运行完成后,在联合分析结果处查看分析完成的报告和结果,示例图如下:
结果解读
微生物和代谢组联合分析从两个层面去进行联合,整体层面去展示微生物和代谢物之间的整体相关性,比如普氏分析、代谢物聚类-菌群相关性分析;从差异层面则基于不同的差异处理组,去进一步联系表型关系,看具体的微生物和代谢之间的关系是否与生物的表型差异相关,这里则主要有差异代谢物和菌群相关性分析、协惯量分析、典范对应分析、随机森林分析等,接下来我们就这些分析和大家具体看下怎么解读和应用。
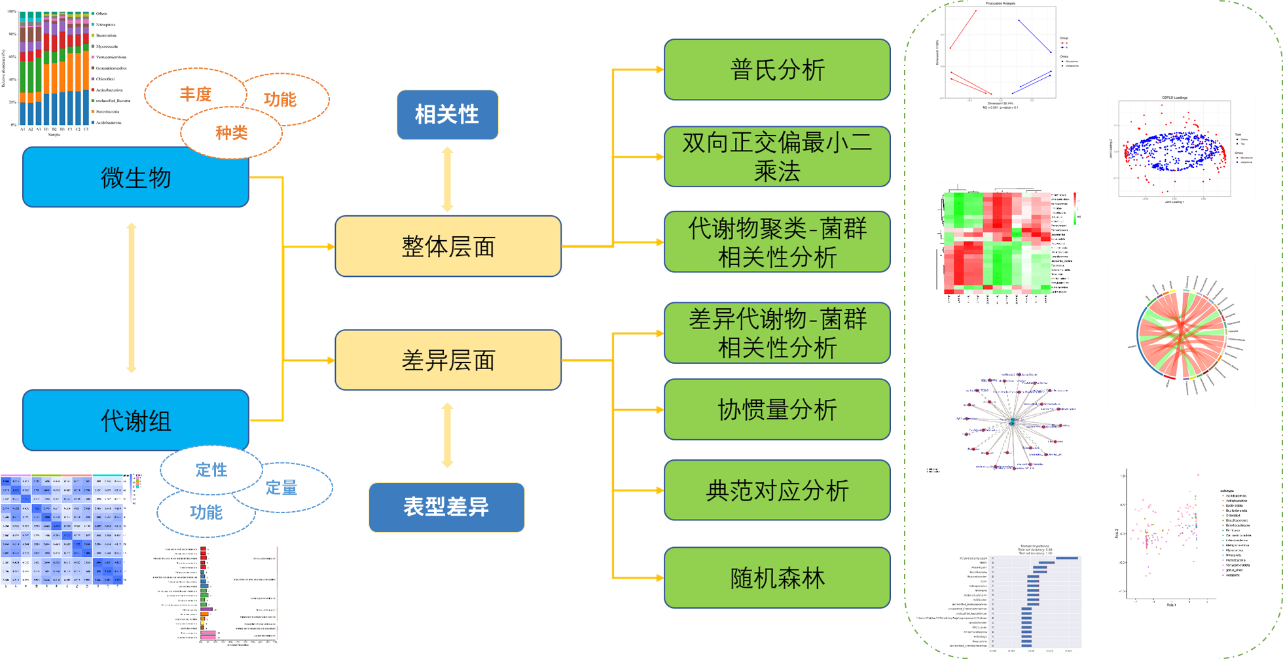
整体分析流程图
整体层面
1、普氏分析
普氏分析(Procrustes analysis)是一种多元统计学中的形状比较分析的方法,即通过分析形状分布,比较两组数据一致性。普氏分析是基于匹配两个数据集中的对应点(坐标),通过平移、旋转和缩放其中一个数据集中点的坐标以匹配另一数据集中对应点的坐标,并最小化点坐标之间的偏差平方和(表示为M2)。对应点坐标之间的偏差称为矢量残差(vector residuals),越小的矢量残差代表了两数据集具有更高的一致性。
由于两组数据集的属性不同,并不适合用来直接比较,可分别对两个数据集降维排序分析,并提取特征轴的坐标。本分析选用 PCoA 分别对微生物组(属水平)和代谢组进行降维排序,之后进行Procrustes分析,比较微生物组与代谢组之间的相似和变异情况。
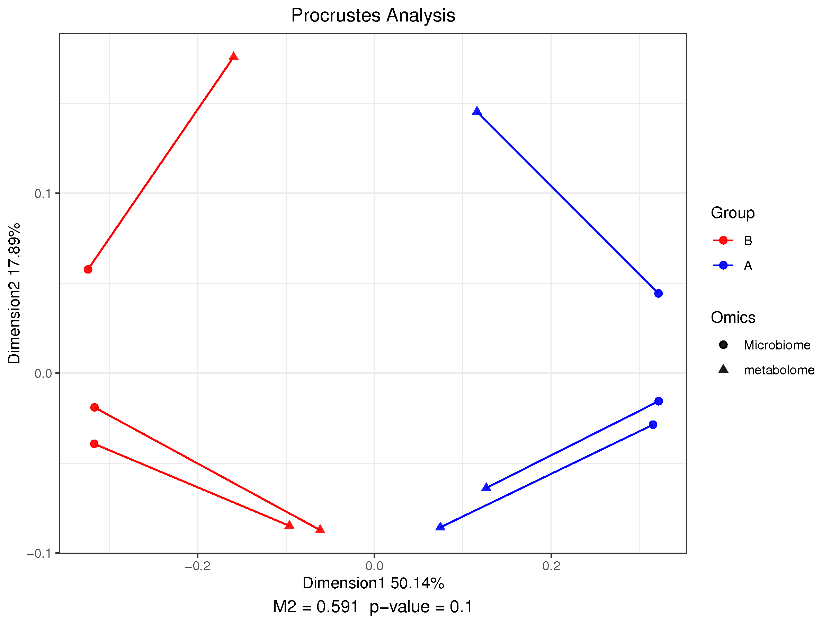
普氏分析结果图
注:图中不同颜色代表不同的分组;每个线段代表一个样本,线段一端实心圆点代表微生物组数据样本点,线段另一端实心三角形代表相同样本的代谢组数据样本点;连线代表两排序构型的矢量残差,可评价二者间的变异情况,连线越短,表示两个数据集之间一致性越高。Monte Carlo Label Permutations P 表示由 Monte Carlo 算法模拟生成的 p-value,p
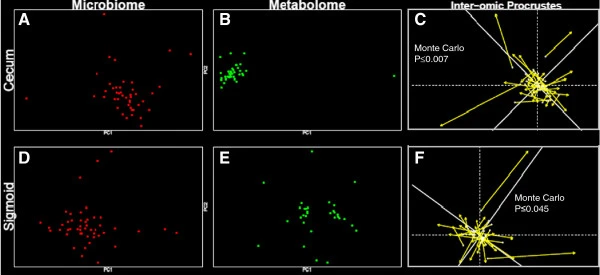
文章案例
图注:Procrustes分析:第一列图表包含微生物组数据(红点),第二列包含代谢组数据(绿点)。第一行包含盲肠数据,第二行包含乙状结肠数据。对盲肠微生物组(A)、盲肠代谢组(B)、乙状结肠微生物组(D)和乙状结肠代谢组(E)进行主成分分析。然后进行样本点间(C和F) Procrustes分析。Procrustes图上的线越长,表明微生物组和代谢组的对象内差异越大。所示的显著性值是使用Vegan R软件包中的protest函数计算的,该函数进行重复的对称Procrustes分析以估计显著性。
引用:McHardy I H, Goudarzi M, Tong M, et al. Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships[J]. Microbiome, 2013, 1(1): 1-19.
2、双向正交偏最小二乘法(O2PLS)
双向正交偏最小二乘法(Two-way orthogonal partial least squares,O2PLS)通过对两个数据组间的整合分析,评估两个数据集之间的内在相关性。两组数据是否可建立O2PLS模型,可判断数据组间是否存在关联性。O2PLS模型一方面可反映不同数据组间的整体影响,另一方面可直接体现不同变量在模型中的权重(权重越大,意味着该变量的变化对另一个组学的扰动更剧烈),从而更加精准地发现关键调节现象。O2PLS 为非监督建模,可客观描述两数据组间是否存在关联趋势,尽可能从源头上避免假阳性关联。该方法先对菌落和代谢的数据进行UV scaling预处理,再构建群落和代谢O2PLS模型,计算每个样本的得分,得到联合得分图;然后计算每种微生物和代谢物的载荷值,得到载荷图。联合得分图指示了两个数据矩阵之间的关系,具有高载荷值的代谢物/微生物被认为是两个数据集相似性所必需的。最后选择前两个维度载荷值长度 top15 代谢物/微生物绘制柱状图。
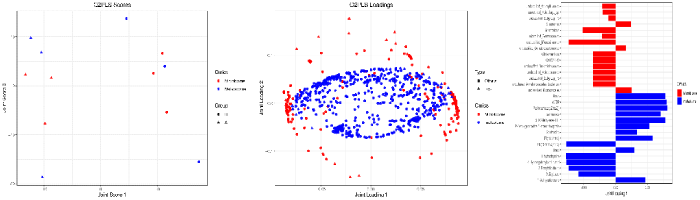
O2PLS 联合样本得分图(左)、O2PLS 模型微生物和代谢物的载荷图(中)、O2PLS 关联程度大的微生物/代谢物柱状图(右)
左图注:图中点的形状(圆点和三角形)代表不同分组的样本,点的颜色代表不同的组学样本,红色代表微生物组样本,蓝色表示代谢物样本,图中横纵坐标表示代谢组和微生物组联合的得分。
中图注:图中点的颜色代表不同组学数据,红色代表微生物,蓝色代表代谢物。横纵坐标表示联合载荷值。载荷值长度 top15 代谢物/微生物表示为Top,用实心三角形表示,其它的代谢物/微生物表示为Others,用实心圆点表示。
右图注:图中展示了载荷值长度 top10 的代谢物和载荷值长度 top15 的微生物,红色柱子代表微生物,蓝色柱子代表代谢物。横坐标表示联合载荷轴 1,纵坐标表示代谢物/微生物。
3、代谢物聚类-微生物相关性分析
相关性分析(correlation analysis)是指对两个或多个具备相关性的变量元素进行分析,从而衡量两个变量因素的相关密切程度。基于皮尔逊相关系数,可以度量环境样本中物种多样性与代谢物之间的关联程度。从整体水平对代谢物和菌群进行相关性分析,相关性分析结果以热图,和弦图,网络图三种形式展现。
(1)代谢物簇-微生物相关性热图:一种代谢物簇/微生物可能与多种微生物/代谢物簇具有相关性,保留至少含一组相关性的pvalue 满足 CCP<0.05 的数据,然后进行热图的绘制。热图可以通过颜色梯度来反应各种代谢物与各个微生物分类之间的相关性大小,并且可以对多样性和代谢物进行聚类分析。
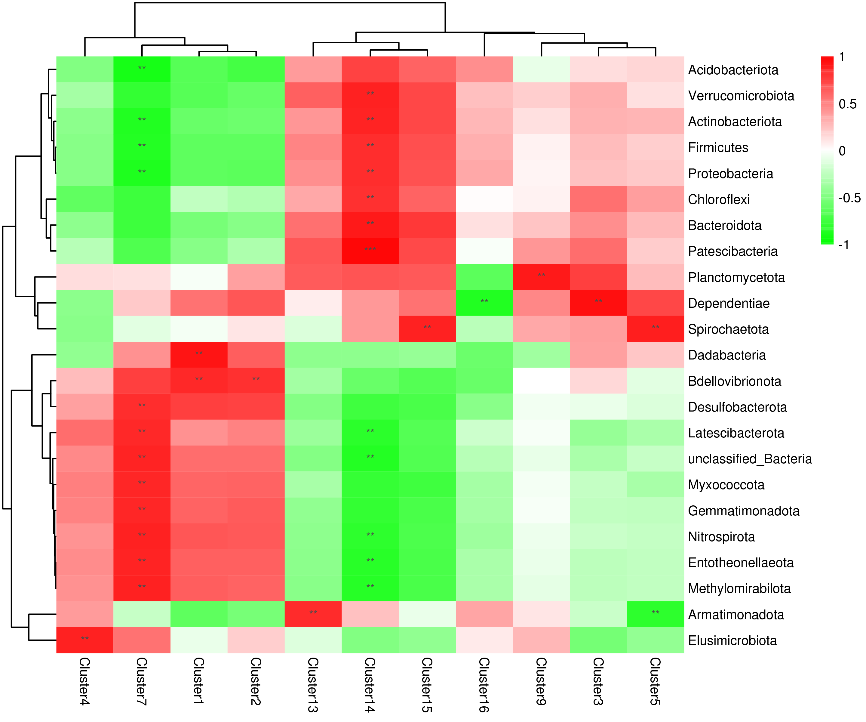
代谢物簇-微生物相关性热图
注:图右侧为微生物(门水平),下方为代谢物簇,左侧和上方分别为微生物和代谢物簇的聚类树状图。不同颜色代表皮尔逊相关系数的大小,绝对值越接近于 1,相关性越高。红色表示正相关,而绿色表示负相关。星号表示代谢物簇和微生物之间显著性相关(p<0.05),*、**和***表示相关性显著程度依次增强,分别表示 p<0.05,p<0.01和 p<0.001.
(2)代谢物簇-微生物和弦图:将top30 频数的代谢物簇/微生物的相关性结果表,保留至少含一组相关性系数绝对值在Top 30(按照相关系数绝对值从大到小排序)内的数据 ,用于相关性弦图的绘制。
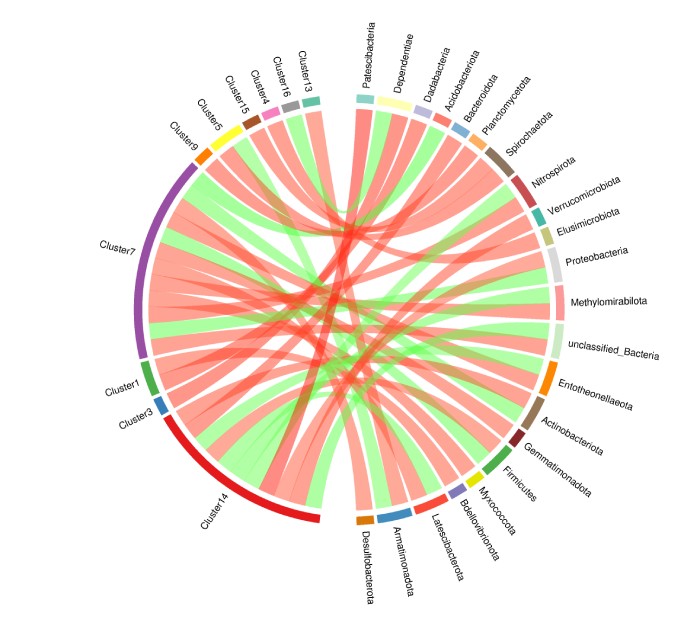
代谢物簇-微生物相关性和弦图
注:弦图有两种形式:文件名含label的和弦图,代谢物簇/微生物名称在圈外展示;文件名含legend的和弦图,代谢簇在左下角展示,微生物在右下角展示。弦图左半圈为代谢物,右半圈为微生物,每一条弦表示该代谢物与该微生物具有显著相关性,红色弦代表正相关,绿色弦代表负相关。弦的宽度越宽,表示与这个代谢物或微生物相关的频数 count 越多。
(3)代谢物簇-微生物网络图:利用代谢物簇-微生物和弦图的相关性系数表,用于相关性网络图的绘制。
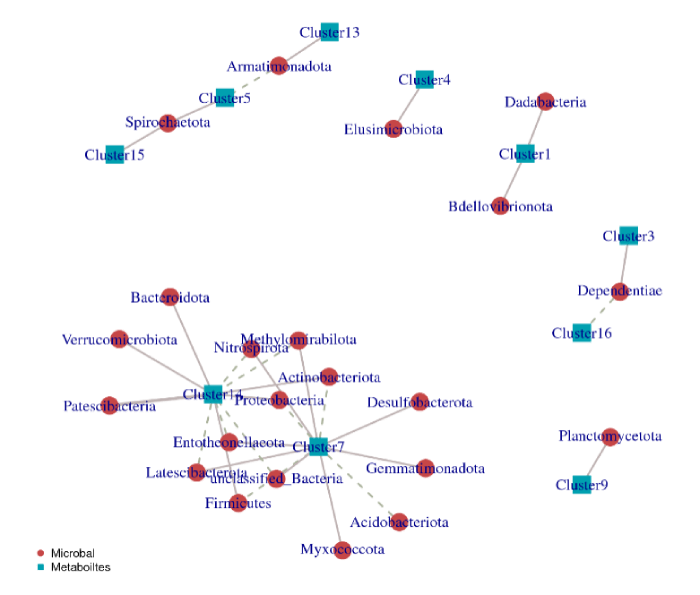
代谢物簇-微生物相关性网络图
注: 图中代谢物簇用蓝色方形标出,微生物用红色圆形标出。实线代表正相关,虚线代表负相关。
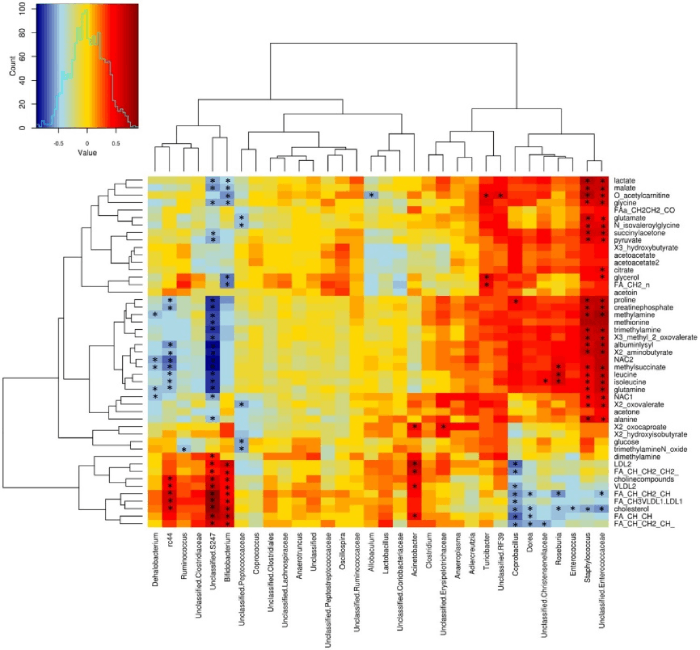
文章案例:结肠微生物组和血清代谢组之间的联系
热图显示了ND组和3xtg组的代谢物概况和特定细菌家族和属的相对丰度之间的关联。红到蓝的比例:从正到负的关联。Pearson’s correlations与数据分布一致,经Shapiro-Wilk测试验证。*p < 0.05.
引用:Sanguinetti E, Collado M C, Marrachelli V G, et al. Microbiome-metabolome signatures in mice genetically prone to develop dementia, fed a normal or fatty diet[J]. Scientific Reports, 2018, 8(1): 1-13.
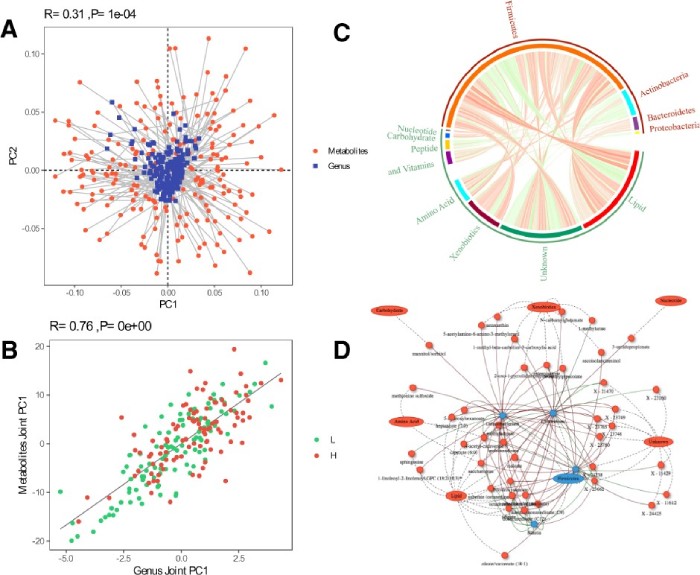
相似性分析和spearman相关分析结果的说明
(A) Procrustes analysis (PA):连接两点的线的长度表示两个数据集之间样本的一致性。(B) O2PLS模型:O2PLS模型中具有较大的荷载值,被认为是它们相似性的重要因素。(C) 微生物和代谢物之间的spearman相关性的Circos图。(D) 不同的代谢物和属于韧皮部的微生物之间的spearman相关网络。
引用:Ni Y, Yu G, Chen H, et al. M2IA: a web server for microbiome and metabolome integrative analysis[J]. Bioinformatics, 2020, 36(11): 3493-3498.
差异层面
1、差异代谢物-菌群相关性分析
基于差异代谢物和差异菌群(genus 属水平)进行相关性分析,不对差异代谢物进行降维处理,其他分析原理同整体层面的代谢物聚类-微生物相关性分析。也是以相关性热图,和弦图,网络图三种形式展现。
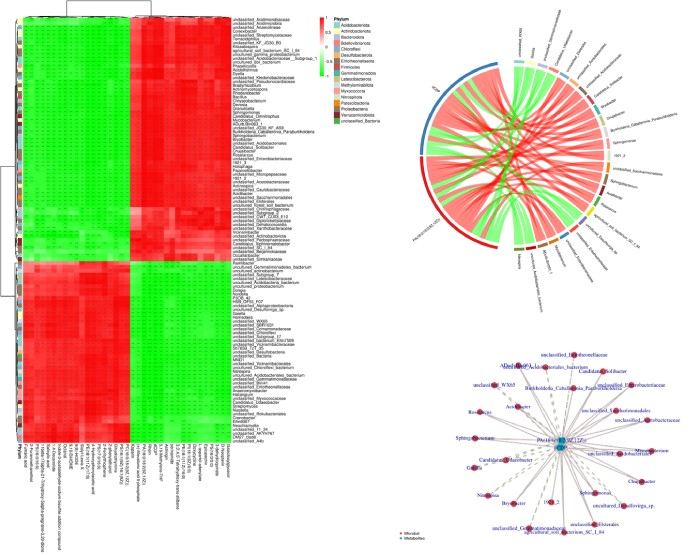
差异代谢物-差异菌群相关性热图(左)、差异代谢物-差异菌群相关性和弦图(右上)、差异代谢物-差异菌群相关性网络图(右下)
2、协惯量分析
协惯量分析(coinertia analysis)可用于两组变量的分析,常见于生态学中研究植被与环境的关系,随后被运用到多组学联合分析中。选择差异微生物(属水平),并将微生物将按照门划分类,结合差异代谢物可展现出差异代谢物和微生物(属水平)之间的关系,也能体现微生物不同类群间的分布情况,一般情况下同类群的微生物分布相对集中,不同的差异分组中的同类微生物分布可能不同。
3、典范对应分析
典范对应分析(canonical correspondence analysis, CCA),是基于对应分析发展而来的一种排序方法,将对应分析与多元回归分析相结合,每一步计算均与环境因子进行回归,又称多元直接梯度分析。将差异代谢物视为环境因子,与差异菌群(属水平)进行典型对应分析能揭示菌群的分布特点的同时也能挖掘与之相关联的代谢物。
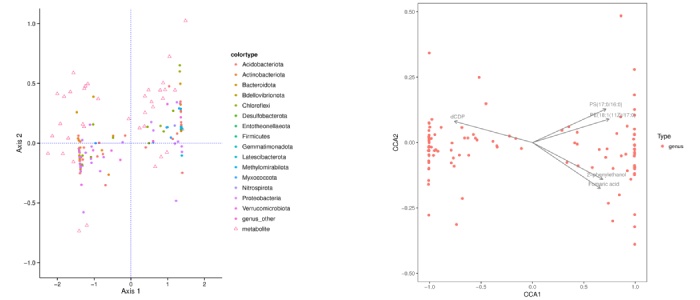
协惯量分析结果图(左)、典范对应分析分析结果图(右)
左图注:图中一个圆代表一个微生物,不同的颜色表示不同的分类(按门划分,按照门名称排序,前15个门显示对应门名称,其他门通称为*_other),三角形是代谢物;微生物、代谢物与原点连线形成的夹角反映了代谢物和微生物之间的相关性,锐角为正相关,钝角为负相关,直角为不相关。
右图注:图中的点表示微生物,箭头为代谢物(为了更清晰的展示代谢物,根据图幅,统一将箭头的长度进行了一定比例的缩放),仅展示箭头长度top5 的代谢物名称。微生物和原点连线与箭头形成的夹角反映了代谢物和微生物之间的相关性,锐角为正相关,钝角为负相关,直角为不相关。
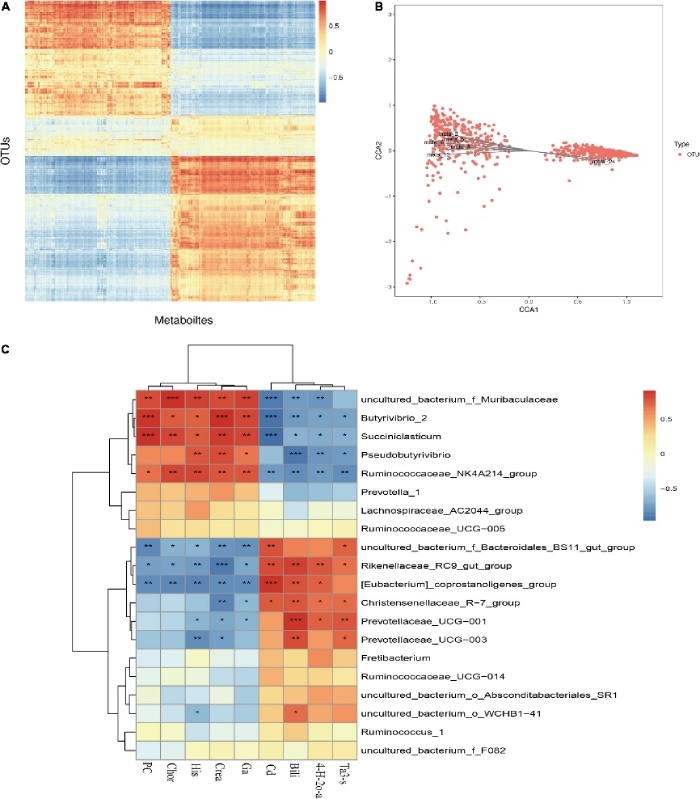
文章案例:瘤胃微生物和代谢物的相关性分析
(A) 差异代谢物和OTU之间的相关性分析。(B) 差异代谢物和微生物之间的限制性对应分析。(C) 差异代谢物和微生物区系之间的相关热图。*P < 0.05; **P < 0.01; ***P < 0.001.
引用:Liu X, Sha Y, Lv W, et al. Multi-omics reveals that the rumen transcriptome, microbiome and its metabolome co-regulate cold season adaptability of Tibetan sheep[J]. Frontiers in microbiology, 2022: 887.
4、随机森林分析
在机器学习中,随机森林(Random Forest)是一个含多个决策树的分类器,并且其输出的类别是由个别树输出的类别的众数而定。在随机森林中,集成模型中的每棵树构建时的样本都是由训练集经过有放回抽样得来的。随机森林构建过程的随机性能够产生具有不同预测错误的决策树。通过取这些决策树的平均,能够消除部分错误。随机森林建模可以在分类时评估特征的重要性。随机森林建模被广泛应用于少样本、高特征维度的数据集建模中。在多组学联合分析中,可以通过对模型特征的排序筛选出对模型重要的特征,从而起到筛选biomarker的目的。还可以通过不同组学的单独建模与合并数据建模的ROC曲线(受试者回归曲线,Receiver Operating Characteristic curve)对比,评估哪种组学能更好地分离对照组和实验组。
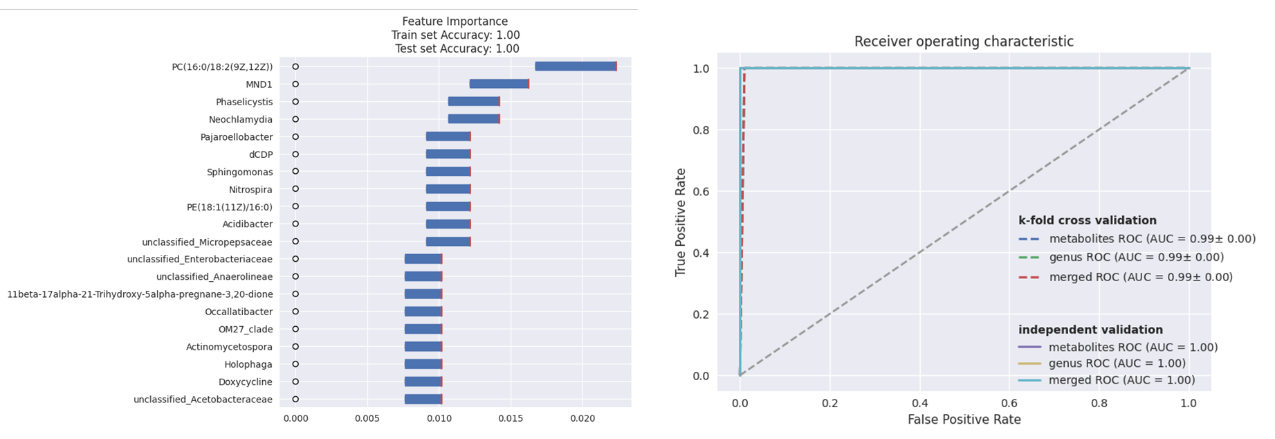
随机森林分类器特征重要性排序箱线图(左)、随机森林分类器ROC曲线图(右)
左图注:将两组数据合并建模并按模型特征重要性从大到小排序,图片显示top 20的特征,依据K重交叉验证结果作箱线图。
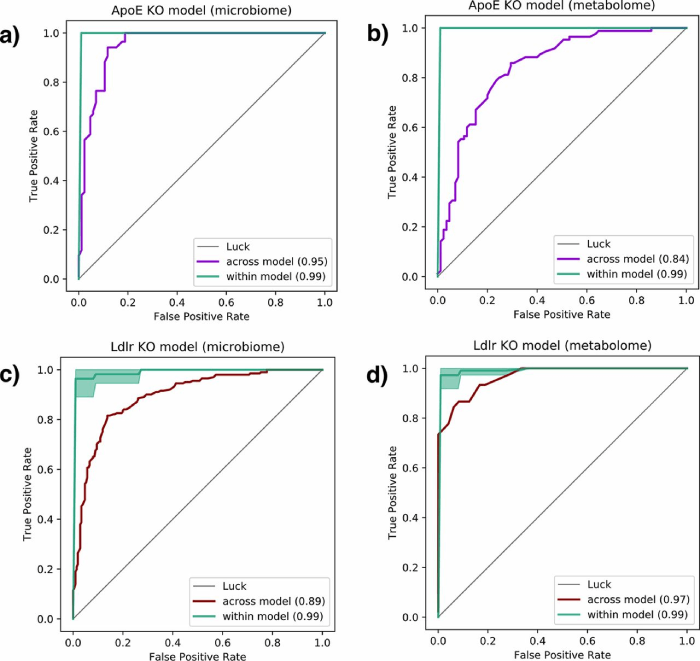
文章案例:使用随机森林模型评估预测IHH暴露能力的ROC曲线
右图注:虚线显示训练集K重交叉验证的平均ROC曲线,实线显示独立验证的ROC曲线。不同颜色虚线和实线分别表示代谢组、微生物组(属水平)分别建模以及合并建模的ROC曲线。
绿色曲线代表每个小鼠模型内的分类准确性。紫色ROC曲线对应于使用来自ApoE-/-小鼠模型的肠道微生物组(a)和代谢组(b)数据训练的模型,以预测Ldlr-/-小鼠的IHH暴露。红色曲线显示的是在ApoE-/-小鼠上测试的Ldlr-/-小鼠的微生物组(c)和代谢组(d)数据。IHH:间歇性低氧和高碳酸血症。
引用:Tripathi A, Xu Z Z, Xue J, et al. Intermittent hypoxia and hypercapnia reproducibly change the gut microbiome and metabolome across rodent model systems[J]. MSystems, 2019, 4(2): e00058-19.





 京公网安备 11011302003368号
京公网安备 11011302003368号