

今天给大家介绍的是BMKMANU S1000空间转录组数据与单细胞转录组数据的关联分析。在我们的关联分析中可以使用不同分辨率的空间数据分别与单细胞数据进行关联分析,本期我们使用RCTD进行关联分析。详细分析流程如下所示:
RCTD
使用监督学习的方法,利用带注释的scRNA-seq数据来定义空间转录组学数据中预期存在的细胞类型的细胞特异性情况。
1、单细胞数据格式要求
(1)data 1 细胞注释结果(cellType)
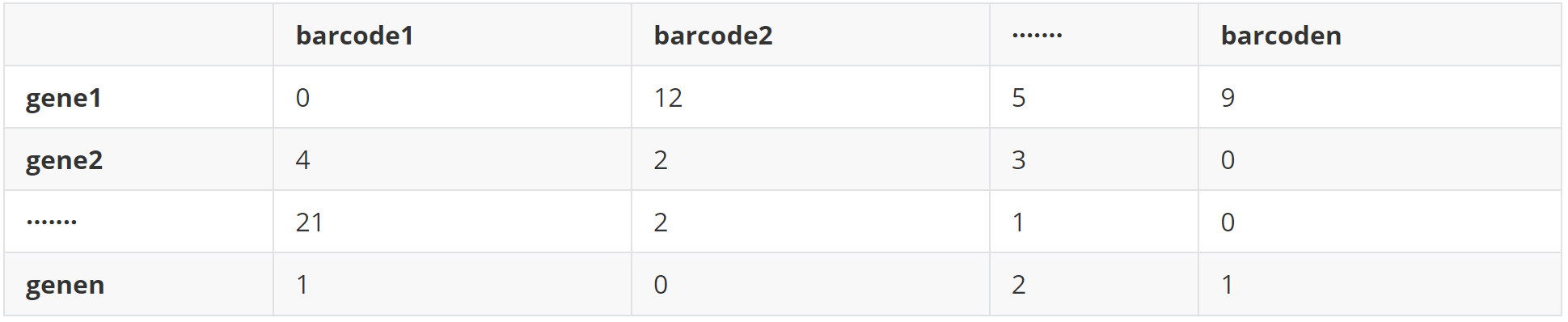
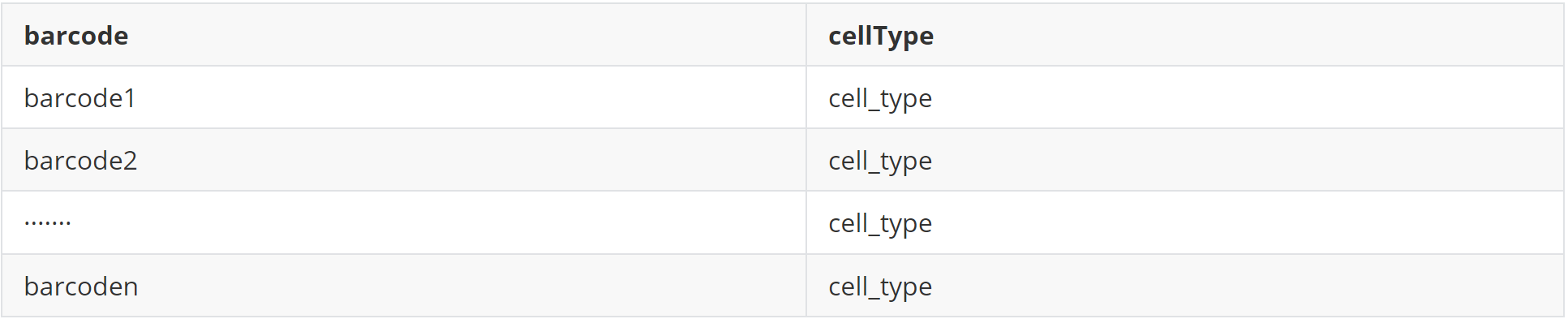 (2)data 2 基因表达矩阵(sc_counts)
(2)data 2 基因表达矩阵(sc_counts)
 2、空间数据格式要求
2、空间数据格式要求
(1)data 1 空间spots位置信息(coords)
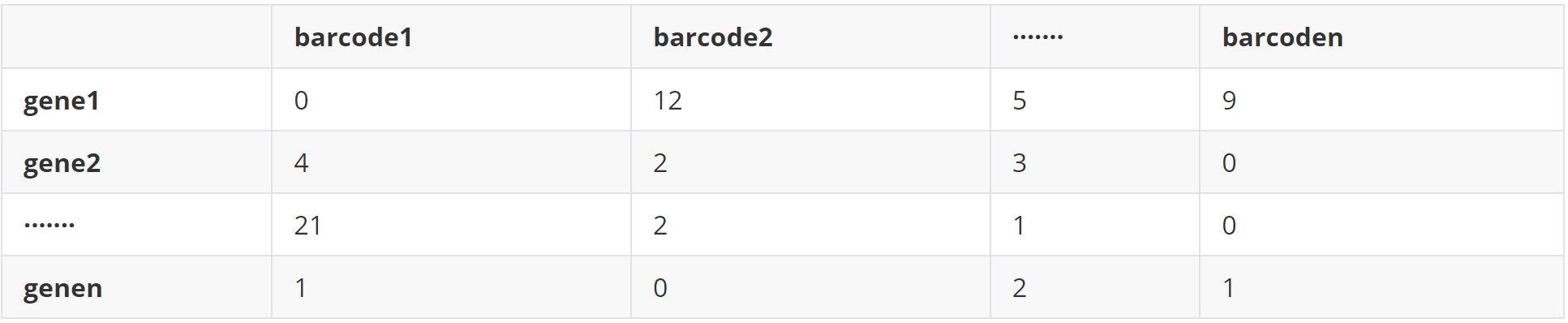
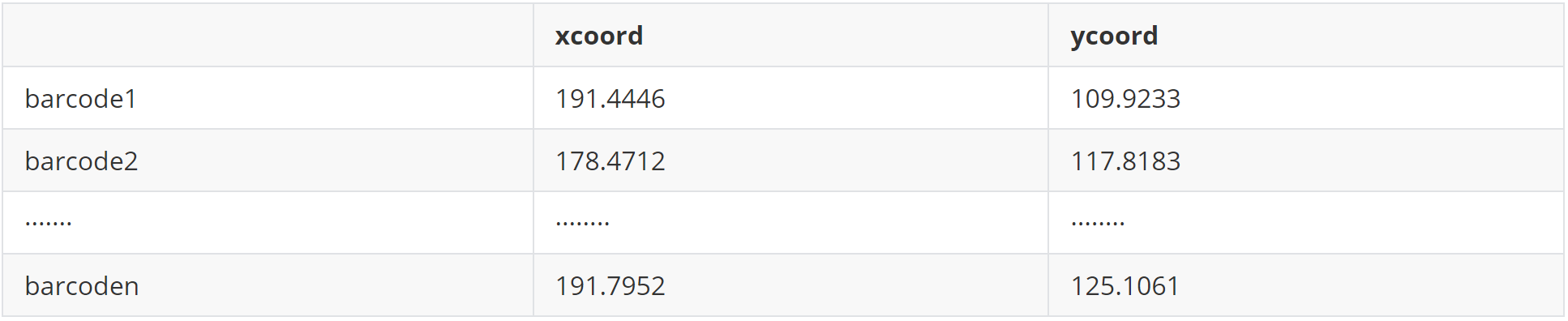 (2)data 2 基因表达矩阵(sp_counts)
(2)data 2 基因表达矩阵(sp_counts)
 #安装spacexr包
#安装spacexr包
# install.packages(“devtools”)
devtools::install_github(“dmcable/spacexr”, build_vignettes = FALSE)
#加载R包
library(Seurat)
library(tidyverse)
library(Matrix)
library(spacexr)
library(ggplot2)
library(ggpubr)
library(gridExtra)
library(reshape2)
library(readr)
library(Seurat)
library(config)
library(ggpubr)
library(gridExtra)
library(reshape2)
library(png)
library(patchwork)
library(SingleR)
library(celldex)
#矩阵转化
Rcpp::sourceCpp(code=’
#include <Rcpp.h>
using namespace Rcpp;
// [[Rcpp::export]]
IntegerMatrix asMatrix(NumericVector rp,
NumericVector cp,
NumericVector z,
int nrows,
int ncols){
int k = z.size() ;
IntegerMatrix mat(nrows, ncols);
for (int i = 0; i < k; i++){
mat(rp[i],cp[i]) = z[i];
}
return mat;
}
‘ )
as_matrix <- function(mat){
row_pos <- mat@i
col_pos <- findInterval(seq(mat@x)-1,mat@p[-1])
tmp <- asMatrix(rp = row_pos, cp = col_pos, z = mat@x,
nrows = mat@Dim[1], ncols = mat@Dim[2])
row.names(tmp) <- mat@Dimnames[[1]]
colnames(tmp) <- mat@Dimnames[[2]]
return(tmp)
}
#读取单细胞数据
expr <- Read10X(‘/xxx/04.QC/filtered_feature_bc_matrix/’, cell.column = 1)
sc_data <- CreateSeuratObject(counts = expr,assay = “RNA”,min.cells=5,min.features=100)
#counts & nUMI
sc_counts <- as_matrix(sc_data[[‘RNA’]]@counts)
sc_nUMI = colSums(sc_counts)
#注释
##方法一:SingleR 注释
load(‘/share/nas1/guochao/database/celldex/MouseRNAseqData.Rdata’)
mouseRNA <- ref
sce_for_SingleR<-GetAssayData(sc_data,slot=”data”)
pred.mouseRNA <- SingleR(test=sce_for_SingleR,ref=mouseRNA,labels=mouseRNA$label.fine,assay.type.test=”logcounts”,assay.type.ref =”logcounts”)
pred.mouseRNA$labels<-as.factor(pred.mouseRNA$labels)
cellType=data.frame(barcode=sc_data@assays$RNA@counts@Dimnames[2], celltype=pred.mouseRNA$labels)
names(cellType) = c(‘barcode’, ‘cell_type’)
cell_types = cellType$cell_type; names(cell_types) <- cellType$barcode # create cell_types named list
cell_types = as.factor(cell_types) # convert to factor data type
##方法二:手动注释
sc_data = NormalizeData(sc_data, normalization.method = ‘LogNormalize’, scale.factor = 10000)
sc_data = FindVariableFeatures(sc_data, selection.method = ‘vst’, nfeatures = 2000)
sc_data = ScaleData(sc_data)
sc_data = RunPCA(sc_data, features = VariableFeatures(object = sc_data))
sc_data = FindNeighbors(sc_data, dims = 1:15)
sc_data = FindClusters(sc_data, resolution = 0.25)
markers = FindAllMarkers(sc_data, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
top10 = markers %>% group_by(cluster) %>% top_n(n = 5, wt = avg_log2FC)
my_df = sc_data@meta.data %>% as.data.frame() %>%
select(seurat_clusters) %>%
rownames_to_column(var = ‘barcode’) %>%
rename(cluster = seurat_clusters)
cellType = my_df %>% mutate(cell_type = case_when( cluster == ‘0’ ~ ‘Allantois cell’,
cluster == ‘1’ ~ ‘Bergmann glial cell’,
cluster == ‘2’ ~ ‘Neuroblast’,
cluster == ‘3’ ~ ‘Neuroendocrine cell’,
cluster == ‘4’ ~ ‘Fibroblast’,
cluster == ‘5’ ~ ‘Type I spiral ganglion neuron’,
cluster == ‘6’ ~ ‘Erythroid cell’,
cluster == ‘7’ ~ ‘Type II spiral ganglion neuron’,
cluster == ‘8’ ~ ‘Epithelial cell’,
cluster == ‘9’ ~ ‘Cardiac progenitor cell’,
cluster == ’10’ ~ ‘Oligodendrocyte precursor cell’,
cluster == ’11’ ~ ‘Endothelial cell’,
cluster == ’12’ ~ ‘Megakaryocyte’,
cluster == ’13’ ~ ‘Hepatocyte’,
cluster == ’14’ ~ ‘Ventricular cardiomyocyte’))
cell_types = cellType$cell_type; names(cell_types) <- cellType$barcode # create cell_types named list
cell_types = as.factor(cell_types) # convert to factor data type
#构建参考集
reference = Reference(sc_counts, cell_types, sc_nUMI)
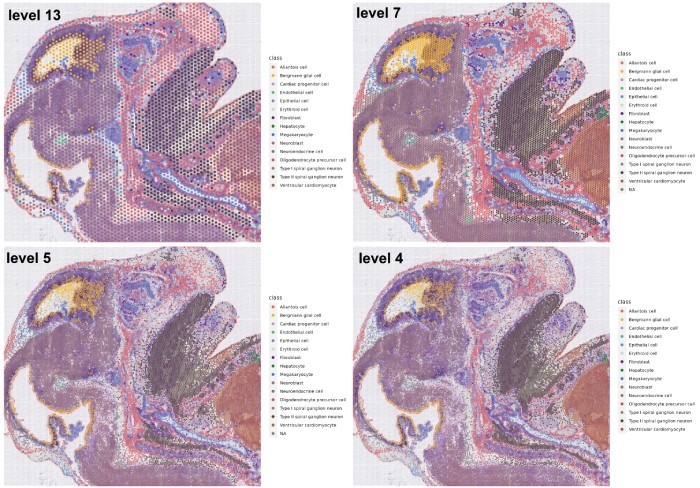
#读取空间数据,这里选择的是level 13的数据
#位置信息
coords = read.table(gzfile(‘/xxx/05.AllheStat/BSTViewer_project/subdata/L13_heAuto/barcodes_pos.tsv.gz’), header = F) %>% dplyr::rename(barcodes = V1, xcoord = V2, ycoord = V3)
rownames(coords) <- coords$barcodes; coords$barcodes <- NULL
#表达量矩阵
expr <- Read10X(‘/xxx/05.AllheStat/BSTViewer_project/subdata/L13_heAuto/’, cell.column = 1)
sp_data <- CreateSeuratObject(counts = expr,assay = “Spatial”)
sp_counts <- as_matrix(sp_data[[‘Spatial’]]@counts)
#nUMI
sp_nUMI <- colSums(sp_counts)
#构建空间实验集
puck <- SpatialRNA(coords, sp_counts, sp_nUMI)
#联合分析
myRCTD <- create.RCTD(puck, reference, max_cores = 8)
myRCTD <- run.RCTD(myRCTD, doublet_mode = ‘doublet’) #run.RCTD
#查找marker基因
get_marker_data <- function(cell_type_names, cell_type_means, gene_list) {
marker_means = cell_type_means[gene_list,]
marker_norm = marker_means / rowSums(marker_means)
marker_data = as.data.frame(cell_type_names[max.col(marker_means)])
marker_data$max_epr <- apply(cell_type_means[gene_list,],1,max)
colnames(marker_data) = c(“cell_type”,’max_epr’)
rownames(marker_data) = gene_list
marker_data$log_fc <- 0
epsilon <- 1e-9
for(cell_type in unique(marker_data$cell_type)) {
cur_genes <- gene_list[marker_data$cell_type == cell_type]
other_mean = rowMeans(cell_type_means[cur_genes,cell_type_names != cell_type])
marker_data$log_fc[marker_data$cell_type == cell_type] <- log(epsilon + cell_type_means[cur_genes,cell_type]) – log(epsilon + other_mean)
}
return(marker_data)
}
cell_type_info_restr = myRCTD@cell_type_info$info
de_genes <- get_de_genes(cell_type_info_restr, puck, fc_thresh = 3, expr_thresh = .0001, MIN_OBS = 3)
marker_data_de = get_marker_data(cell_type_info_restr[[2]], cell_type_info_restr[[1]], de_genes)
saveRDS(marker_data_de,’/share/nas1/guochao/Test/221107marker_data_de_standard.RDS’)
write.table(marker_data_de, file = ‘/share/nas1/guochao/Test/221107marker_data_de_standard.tsv’, sep = ‘\t’,quote = F, row.names = T)
#构建绘图数据
results <- myRCTD@results
results_df <- results$results_df
barcodes = rownames(results_df[results_df$spot_class != “reject” & puck@nUMI >= 1,])
my_table = puck@coords[barcodes,]
my_table$class = results_df[barcodes,]$first_type
#绘图
cal_zoom_rate <- function(width, height){
std_width = 1000
std_height = std_width / (46 * 31) * (46 * 36 * sqrt(3) / 2.0)
if (std_width / std_height > width / height){
scale = width / std_width
}
else{
scale = height / std_height
}
return(scale)
}
png <- readPNG(‘/share/nas1/dengdj/testing/Barcode_YF/20220923-YF-N1295-1-2/analysis/XSPT-T/05.AllheStat/allhe/he_roi_small.png’)
zoom_scale = cal_zoom_rate(dim(png)[2], dim(png)[1])
my_table = my_table %>% mutate(across(c(x,y), ~.x*zoom_scale))
col = c(“#F56867″,”#FEB915″,”#C798EE”,”#59BE86″,”#7495D3″,”#D1D1D1″,”#6D1A9C”,”#15821E”,”#3A84E6″,”#997273″,”#787878″,”#DB4C6C”,”#9E7A7A”,”#554236″,”#AF5F3C”,”#93796C”,”#F9BD3F”,”#DAB370″)
p = ggplot(my_table, aes(x = x, y = dim(png)[1] – y))+
background_image(png)+
geom_point(shape = 16, size = 1.8, aes(color = class))+
coord_cartesian(xlim = c(0, dim(png)[2]), y = c(0, dim(png)[1]), expand = FALSE)+
scale_color_manual(values = col)+
theme(axis.title.x=element_blank(),axis.text.x=element_blank(),axis.ticks.x=element_blank(), axis.title.y=element_blank(),axis.text.y=element_blank(),axis.ticks.y=element_blank())+
guides(color = guide_legend(override.aes = list(size = 2.5, alphe = 0.1)))
ggsave(p, file = ‘/share/nas1/guochao/Test/temp/221107_all.png’, width=10, height=7, dpi = 300)
ggsave(p, file = ‘/share/nas1/guochao/Test/temp/221107_all.pdf’, width=10, height=7)




 京公网安备 11011302003368号
京公网安备 11011302003368号