
细胞通讯分析流程与方法:细胞通讯分析的基础是基因表达数据和配体-受体数据库信息,例如转录组数据表明A、B细胞分别表达了基因α和β,通过数据库查询α和β是配体-受体关系,则认为A-B通过α-β途径进行了通讯。
输入数据格式要求:
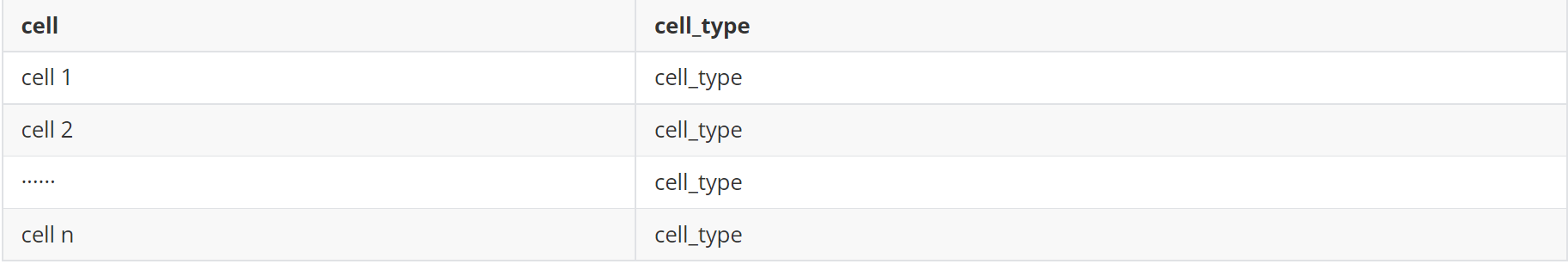
(1)细胞注释文件(meta.txt,两列,分别为细胞id和细胞类型)
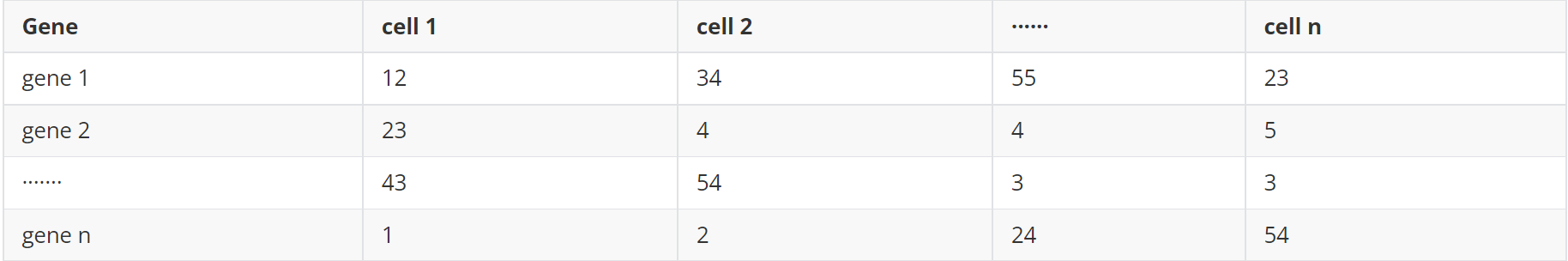
 (2)基因表达矩阵(counts.txt)
(2)基因表达矩阵(counts.txt)
 #下载软件(按顺序安装)
#下载软件(按顺序安装)
conda create -n cellphonedb2 python=3.7
conda install Cython
conda install h5py
conda install scikit-learn
conda install r
conda install rpy2
pip install https://pypi.tuna.tsinghua.edu.cn/simple cellphonedb
pip install -i https://pypi.tuna.tsinghua.edu.cn/simple markupsafe==2.0.1
#R包
install.packages(‘ggplot2’)
install.packages(‘heatmap’)
#data1—> R
#使用RCTD注释的空间数据进行后续分析
results <- myRCTD@results
results_df <- results$results_df
barcodes = rownames(results_df[results_df$spot_class != “reject” & puck@nUMI >= 1,])
my_table = puck@coords[barcodes,]
my_table$class = results_df[barcodes,]$first_type
meta = my_table %>% select(class) %>% rownames_to_column(var = ‘cell’) %>% dplyr::rename(cell_type = class)
write.table(meta ,file = ‘/xxx/Test/meta.txt’, sep = ‘\t’, quote = F, row.names = T)
#data2—> R
expr <- Read10X(‘/xxx/BSTViewer_project/subdata/L13_heAuto/’,cell.column = 1)
object <- CreateSeuratObject(counts = expr,assay = “Spatial”)
counts = object[[‘Spatial’]]@counts %>% as.data.frame() %>% rownames_to_column(var = ‘Gene’)
write.table(counts ,file = ‘/xxx/Test/counts.txt’, sep = ‘\t’, quote = F, row.names = T)
#鼠源基因转化
#CellPhoneDB 只能识别人类基因,如果是鼠源基因需要进行同源基因的转化
#下载人鼠同源基因对应关系
http://asia.ensembl.org/biomart/martview/b9f8cc0248e4714ba8e0484f0cbe4f02
#细胞通讯分析流程与方法参考步骤
https://www.jianshu.com/p/922a7f2c21fc
#下载的对应关系表应该有四列
Gene_stable_ID | Gene_name | Mouse_gene_stable_ID | Mouse_gene_name
#将对应关系(human_mouse_gene.txt)替换到 data2 矩阵—>Shell
awk -F ‘\t’ ‘BEGIN{OFS == ‘\t’}ARGIND==1{a[$4]=$1}ARGIND==2{if(FNR ==1){print}else{$1 = a[$1];print}}’ human_mouse_gene.txt counts.txt | sed ‘/^ /d’ | sed ‘1s/^/Gene\t/’ | sed ‘s/\s/\t/g’ > counts_new.txt
#从远程存储库中列出cellphonedb可用数据库版本:
cellphonedb database list_remote
#查看本地数据库
cellphonedb database list_local
#下载远程数据库
cellphonedb database download –version <version_spec|latest>
#运行cellphonedb —> Shell
cellphonedb method statistical_analysis –output-path test_output meta.txt counts_new.txt
#输出文件
deconvoluted.txt | pvalues.txt | significant_means.txt
test.count_network.txt | means.txt | test.interaction_count.txt
#气泡图—> Shell
cellphonedb plot dot_plot –pvalues-path test_output/pvalues.txt –means-path test_output/means.txt –output-path test_output –output-name test.dotplot.pdf
#仅绘制自己关注的细胞和互作—> Shell
cellphonedb plot dot_plot –pvalues-path test_output/pvalues.txt –means-path test_output/means.txt –output-path test_output –output-name test.dotplot2.pdf –rows test_output/row.txt –columns test_output/col.txt
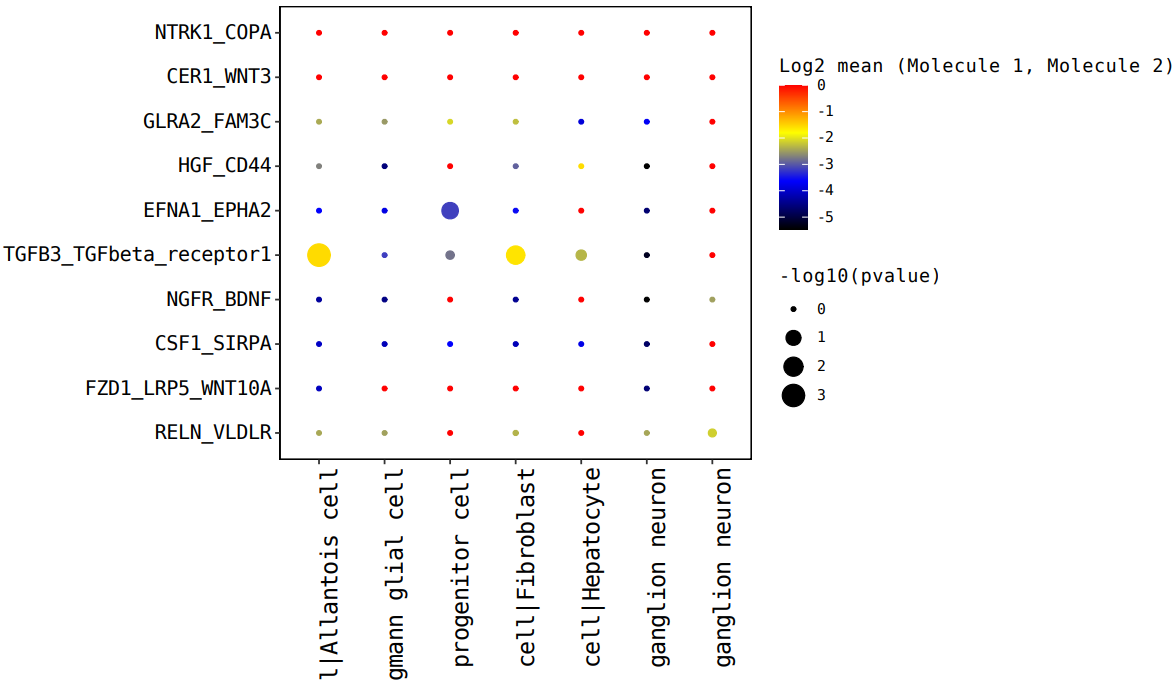
注:横坐标是细胞类型互作,纵坐标是蛋白互作,点越大表示 p 值越小,颜色代表平均表达量
#热图—> Shell
cellphonedb plot heatmap_plot –pvalues-path test_output/pvalues.txt –output-path test_output –pvalue 0.05 –count-name test.heatmap_count.pdf –log-name test.heatmap_log_count.pdf –count-network-name test.count_network.txt –interaction-count-name test.interaction_count.txt meta.txt
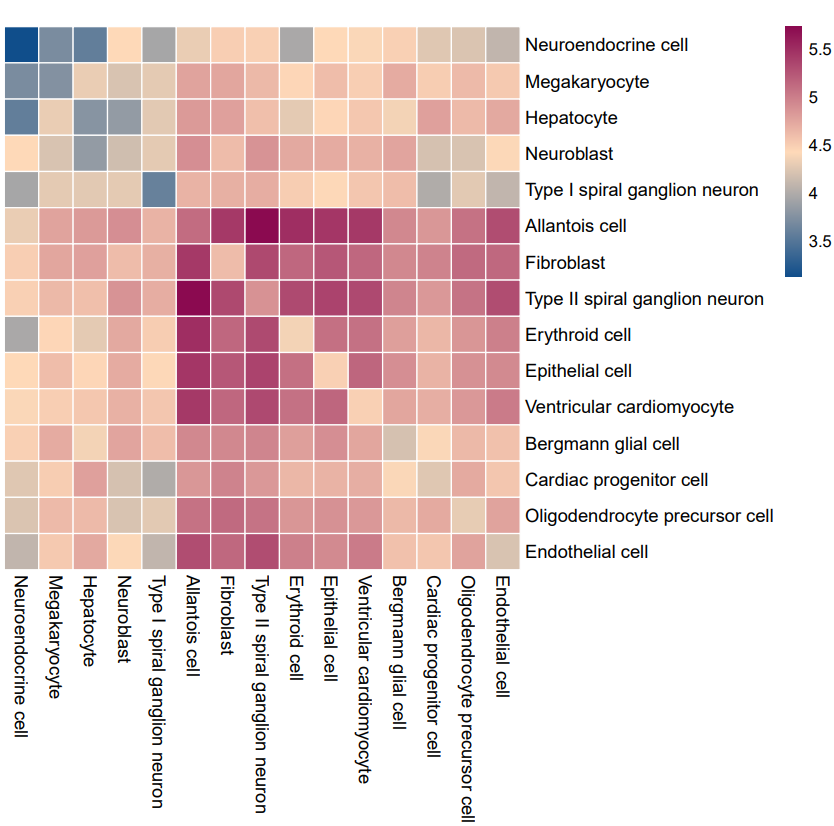
注:横纵坐标表示细胞类型,颜色深蓝色到紫红色代表互作数由低到高
#细胞通讯分析流程与方法:细胞类型网络图—> R
#这里借用另一款细胞通讯分析软件——CellChat(https://github.com/sqjin/CellChat)的绘图方法
#加载R包
library(igraph)
library(tidyr)
library(stats)
library(reshape2)
library(Matrix)
library(grDevices)
library(ggplot2)
#使用CellChat作者提供的绘制互作网络图的function
scPalette <- function(n) {
colorSpace <- c(‘#E41A1C’,’#377EB8′,’#4DAF4A’,’#984EA3′,’#F29403′,’#F781BF’,’#BC9DCC’,’#A65628′,’#54B0E4′,’#222F75′,’#1B9E77′,’#B2DF8A’,
‘#E3BE00′,’#FB9A99′,’#E7298A’,’#910241′,’#00CDD1′,’#A6CEE3′,’#CE1261′,’#5E4FA2′,’#8CA77B’,’#00441B’,’#DEDC00′,’#B3DE69′,’#8DD3C7′,’#999999′)
if (n <= length(colorSpace)) {
colors <- colorSpace[1:n]
} else {
colors <- grDevices::colorRampPalette(colorSpace)(n)
}
return(colors)
}
netVisual_circle <-function(net, color.use = NULL,title.name = NULL, sources.use = NULL, targets.use = NULL, idents.use = NULL, remove.isolate = FALSE, top = 1,
weight.scale = FALSE, vertex.weight = 20, vertex.weight.max = NULL, vertex.size.max = NULL, vertex.label.cex=1,vertex.label.color= “black”,
edge.weight.max = NULL, edge.width.max=8, alpha.edge = 0.6, label.edge = FALSE,edge.label.color=’black’,edge.label.cex=0.8,
edge.curved=0.2,shape=’circle’,layout=in_circle(), margin=0.2, vertex.size = NULL,
arrow.width=1,arrow.size = 0.2){
if (!is.null(vertex.size)) {
warning(“‘vertex.size’ is deprecated. Use `vertex.weight`”)
}
if (is.null(vertex.size.max)) {
if (length(unique(vertex.weight)) == 1) {
vertex.size.max <- 5
} else {
vertex.size.max <- 15
}
}
options(warn = -1)
thresh <- stats::quantile(net, probs = 1-top)
net[net < thresh] <- 0
if ((!is.null(sources.use)) | (!is.null(targets.use)) | (!is.null(idents.use)) ) {
if (is.null(rownames(net))) {
stop(“The input weighted matrix should have rownames!”)
}
cells.level <- rownames(net)
df.net <- reshape2::melt(net, value.name = “value”)
colnames(df.net)[1:2] <- c(“source”,”target”)
# keep the interactions associated with sources and targets of interest
if (!is.null(sources.use)){
if (is.numeric(sources.use)) {
sources.use <- cells.level[sources.use]
}
df.net <- subset(df.net, source %in% sources.use)
}
if (!is.null(targets.use)){
if (is.numeric(targets.use)) {
targets.use <- cells.level[targets.use]
}
df.net <- subset(df.net, target %in% targets.use)
}
if (!is.null(idents.use)) {
if (is.numeric(idents.use)) {
idents.use <- cells.level[idents.use]
}
df.net <- filter(df.net, (source %in% idents.use) | (target %in% idents.use))
}
df.net$source <- factor(df.net$source, levels = cells.level)
df.net$target <- factor(df.net$target, levels = cells.level)
df.net$value[is.na(df.net$value)] <- 0
net <- tapply(df.net[[“value”]], list(df.net[[“source”]], df.net[[“target”]]), sum)
}
net[is.na(net)] <- 0
if (remove.isolate) {
idx1 <- which(Matrix::rowSums(net) == 0)
idx2 <- which(Matrix::colSums(net) == 0)
idx <- intersect(idx1, idx2)
net <- net[-idx, ]
net <- net[, -idx]
}
g <- graph_from_adjacency_matrix(net, mode = “directed”, weighted = T)
edge.start <- igraph::ends(g, es=igraph::E(g), names=FALSE)
coords<-layout_(g,layout)
if(nrow(coords)!=1){
coords_scale=scale(coords)
}else{
coords_scale<-coords
}
if (is.null(color.use)) {
color.use = scPalette(length(igraph::V(g)))
}
if (is.null(vertex.weight.max)) {
vertex.weight.max <- max(vertex.weight)
}
vertex.weight <- vertex.weight/vertex.weight.max*vertex.size.max+5
loop.angle<-ifelse(coords_scale[igraph::V(g),1]>0,-atan(coords_scale[igraph::V(g),2]/coords_scale[igraph::V(g),1]),pi-atan(coords_scale[igraph::V(g),2]/coords_scale[igraph::V(g),1]))
igraph::V(g)$size<-vertex.weight
igraph::V(g)$color<-color.use[igraph::V(g)]
igraph::V(g)$frame.color <- color.use[igraph::V(g)]
igraph::V(g)$label.color <- vertex.label.color
igraph::V(g)$label.cex<-vertex.label.cex
if(label.edge){
igraph::E(g)$label<-igraph::E(g)$weight
igraph::E(g)$label <- round(igraph::E(g)$label, digits = 1)
}
if (is.null(edge.weight.max)) {
edge.weight.max <- max(igraph::E(g)$weight)
}
if (weight.scale == TRUE) {
#E(g)$width<-0.3+edge.width.max/(max(E(g)$weight)-min(E(g)$weight))*(E(g)$weight-min(E(g)$weight))
igraph::E(g)$width<- 0.3+igraph::E(g)$weight/edge.weight.max*edge.width.max
}else{
igraph::E(g)$width<-0.3+edge.width.max*igraph::E(g)$weight
}
igraph::E(g)$arrow.width<-arrow.width
igraph::E(g)$arrow.size<-arrow.size
igraph::E(g)$label.color<-edge.label.color
igraph::E(g)$label.cex<-edge.label.cex
igraph::E(g)$color<- grDevices::adjustcolor(igraph::V(g)$color[edge.start[,1]],alpha.edge)
if(sum(edge.start[,2]==edge.start[,1])!=0){
igraph::E(g)$loop.angle[which(edge.start[,2]==edge.start[,1])]<-loop.angle[edge.start[which(edge.start[,2]==edge.start[,1]),1]]
}
radian.rescale <- function(x, start=0, direction=1) {
c.rotate <- function(x) (x + start) %% (2 * pi) * direction
c.rotate(scales::rescale(x, c(0, 2 * pi), range(x)))
}
label.locs <- radian.rescale(x=1:length(igraph::V(g)), direction=-1, start=0)
label.dist <- vertex.weight/max(vertex.weight)+2
plot(g,edge.curved=edge.curved,vertex.shape=shape,layout=coords_scale,margin=margin, vertex.label.dist=label.dist,
vertex.label.degree=label.locs, vertex.label.family=”Helvetica”, edge.label.family=”Helvetica”) # “sans”
if (!is.null(title.name)) {
text(0,1.5,title.name, cex = 1.1)
}
# https://www.andrewheiss.com/blog/2016/12/08/save-base-graphics-as-pseudo-objects-in-r/
# grid.echo()
# gg <- grid.grab()
gg <- recordPlot()
return(gg)
}
#整体互作网络图绘制
#读取cellphonedb的分析数据
count_net <- read.delim(“/xxx/test_output/test.count_network.txt”, check.names = FALSE)
#数据格式处理
count_inter <- count_net
count_inter$count <- count_inter$count/100
count_inter <- spread(count_inter, TARGET, count)
rownames(count_inter) <- count_inter$SOURCE
count_inter <- count_inter[, -1]
count_inter <- as.matrix(count_inter)
#绘图
netVisual_circle(count_inter,weight.scale = T)
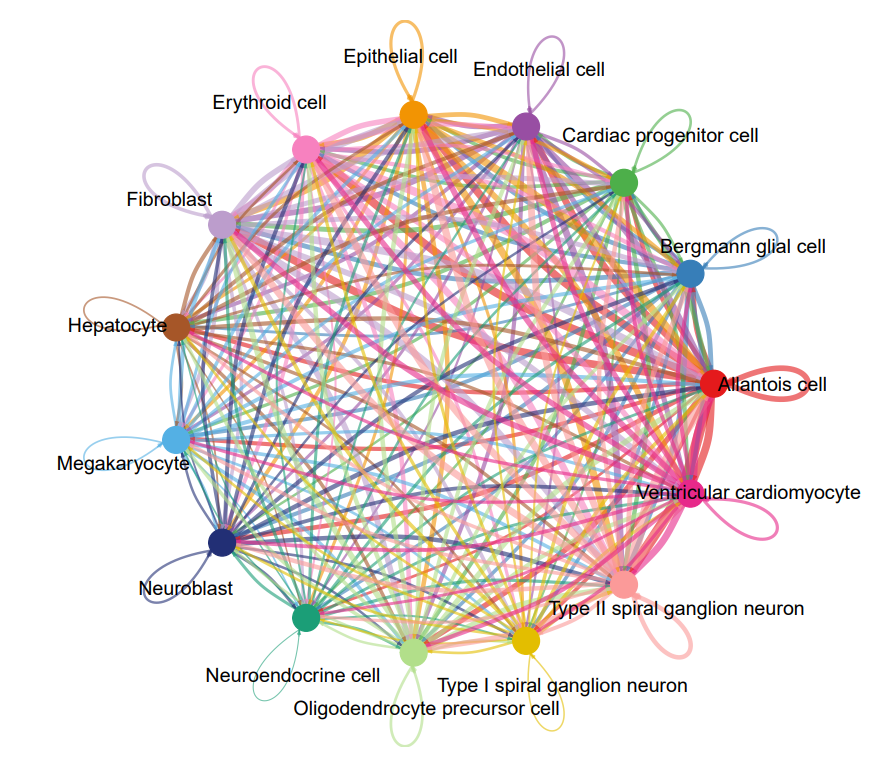
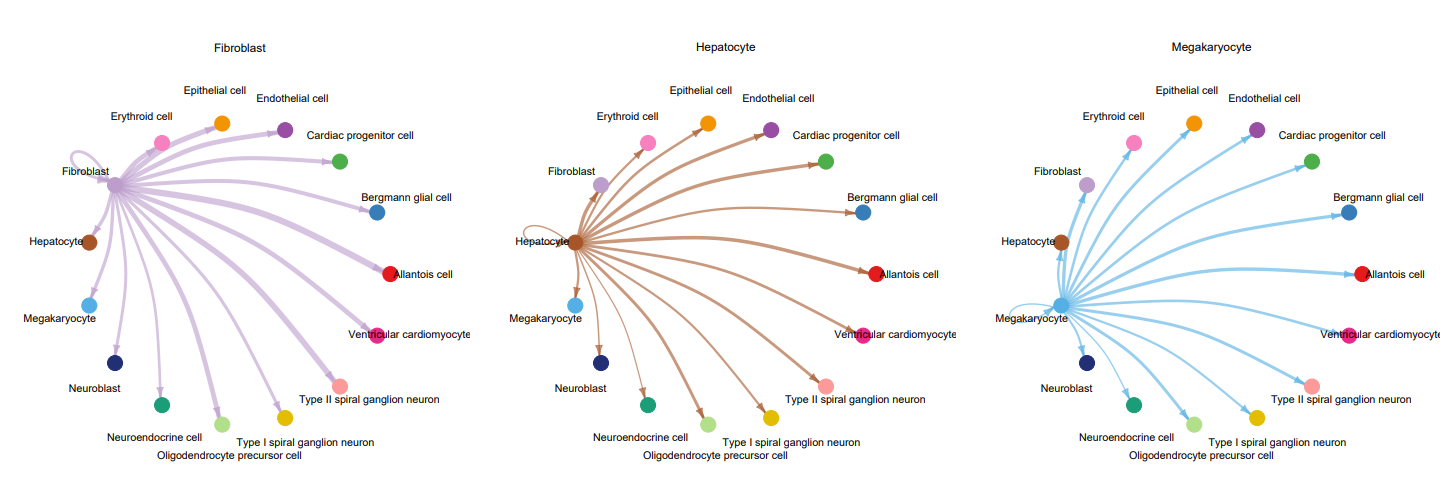
#细胞通讯分析流程与方法:每种细胞与其他细胞的互作
par(mfrow = c(1,3), xpd=TRUE)
for (i in 1:nrow(count_inter)) {
mat2 <- matrix(0, nrow = nrow(count_inter), ncol = ncol(count_inter), dimnames = dimnames(count_inter))
mat2[i, ] <- count_inter[i, ]
netVisual_circle(mat2,
weight.scale = T,
edge.weight.max = max(count_inter),
title.name = rownames(count_inter)[i],
arrow.size=0.2)
}




 京公网安备 11011302003368号
京公网安备 11011302003368号