
四川大学华西第二医院陈路老师团队在《Scientific data》(6.444)在线发表了研究成果。该研究构建了小鼠的长期和短期造血干细胞(HSC)和多能祖细胞(MPP)在批量和单细胞水平上的短读长和长读长RNA测序数据集,数据结果证明了整合短读长和长读长测序可以促进已知和未注释异构体的识别和定量。本文为不同HSC细胞类型内转录多样性和异质性的全面分析和比较研究提供了基础。百迈客为该研究提供了ONT三代长读长测序服务。三代测序平台的转录组研究,无需打断,直接读取反转录的全长cDNA,能够有效的获取高质量的单个RNA分子的全部序列,辨别二代测序无法识别的同源异构体(isoform)、同源基因、超家族基因或等位基因表达的转录本。ONT三代测序其优点有通量更高、操作过程更简单、成本更低,主要应用在基因组测序、甲基化研究、突变鉴定(SNP检测)三个方面。

英文名称:Short-read and long-read RNA sequencing of mouse hematopoietic stem cells at bulk and single-cell levels
中文名称:在批量和单细胞水平上对小鼠造血干细胞进行短读长和长读长RNA测序
发表杂志:Scientific data
影响因子:6.444
发表时间:2021年11月
摘 要
造血干细胞(HSC)位于分化层次的顶端。尽管HSC及其直接下游的多能祖细胞(MPP)具有完全的多向分化能力,但只有长期(LT-)HSC具有长期自我更新的能力。随着单细胞RNA测序和谱系追踪技术的发展,HSC群体内的异质性逐渐得到承认。转录和转录后的调控在控制HSC群体内的分化和自我更新能力方面发挥着重要作用。
在这里本文报告了一个数据集,该数据集包括小鼠长期和短期HSC和MPP在批量和单细胞水平上的短读长和长读长RNA测序。数据结果证明了整合短读长和长读长测序可以促进已知和未注释异构体的识别和定量。因此,该数据集为不同HSC细胞类型内转录多样性和异质性的全面和比较研究提供了基础。
背景介绍
造血始于一群自我更新的造血干细胞 (HSC),它们产生一系列越来越多的谱系定型祖细胞,最终产生各种类型的成熟血细胞。在传统模型中,长期(LT)HCS分化为短期(ST) HSC,随后分化为多能祖细胞(MPP)。虽然这三个群体都具有完全的多向分化能力,但它们逐渐失去了自我更新能力。在HSC和MPP群体中都存在异质性,具有明显的谱系偏差。
转录和转录后的调控在平衡造血干细胞的结构性和低水平周转、下游分化和造血重建方面都是关键。在多细胞生物中,可变剪接是一种关键的转录后调控机制,可以扩大转录本的多样性。越来越多的研究表明,在造血过程中,可变剪接模式是必不可少的。例如,在血液祖细胞或巨核细胞和红细胞谱系中鉴定到的特异性可变剪接事件。研究发现,关键造血调节因子(如HMGA2)的可变剪接模式影响了造血干细胞的分子鉴定。此外,异常AS是包括白血病等各种癌症的标志物。
利用短读长下一代测序(NGS)或长读长测序(如PacBio和Oxford Nanopore Technologies)的RNA测序,是解读包括血细胞生成在内的各种生物过程中的转录多样性和调控机制的强大工具。虽然NGS在表达定量方面更可靠,但是短读长在AS事件中只能提供有限的信息。相比之下,长读长的测序方法提供了一个独特的机会,可以实现在提供全长信息的基础上检测可变剪接异构体。本文使用短读长和长读长RNA测序,在批量和单细胞水平上对小鼠HSC和MPP进行了全面的转录图谱分析。
材料方法
样本制备:8-9周的雌性成年C57BL/6 J 小鼠,从股骨和胫骨中分离骨髓细胞。首先使用小鼠造血干细胞分离试剂盒富集造血干细胞和祖细胞(HSPC)。长期(LT)和短期(ST)造血干细胞(HSC)和多能祖细胞(MPP)根据其表面标志物进行分选。对于单细胞RNA测序(scRNA-seq),将细胞单独分选到含有裂解缓冲液的8条PCR管中。同时对于批量RNA-seq,分选100个细胞(P100)到一个PCR管中作为生物学重复。
实验方法:按照Smart-seq2实验流程构建cDNA 文库,基于Illumina平台、Pacbio平台和Oxford Nanopore Technologies(ONT)(百迈客协助完成该测序服务)平台测序。
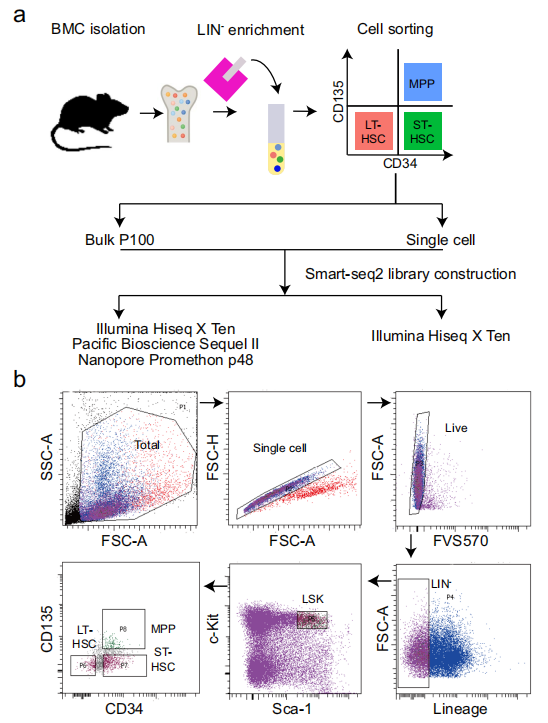
图1 实验设计及样本制备流程
技术验证
- 短读长 Illumina 测序数据的质控
无论是单细胞(图2a)还是批量细胞的水平上(图3a),在不同细胞类型的样本中,每个碱基的平均质量分数分布没有显著差异,并且两个数据集的reads在整个基因体上几乎均匀分布(图2b, 3b),表明RNA的高度完整性。进一步检查了reads被映射到的基因区域,发现所有样本中被映射到外显子区域的reads明显增多,而被映射到内含子区域的reads明显减少(图2c和图3c),与之前的报道结果一致。
对于单细胞测序数据,还检查了映射到线粒体和核糖体基因的reads的比例(图2d)。每个细胞的线粒体基因和细胞核糖体基因的中位数百分比为0.29和3.04。MPP检测到的基因数*高(图2e),显著高于LT-HSC,而每个细胞的UMI数在三种细胞类型之间具有相似性(图2f)。UMAP图表明ST-HSC位于LT-HSC和MPP之间(图2g)。接下来分析细胞类型之间的差异表达基因。LT-HSC、ST-HSC和MPP中分别有62、63和266个差异表达基因。此外一些已知的HSC特征基因,包括Mpl、c-Myc、Mllt3、Gata2,在LT-HSC中表达显著增高(图2h)。
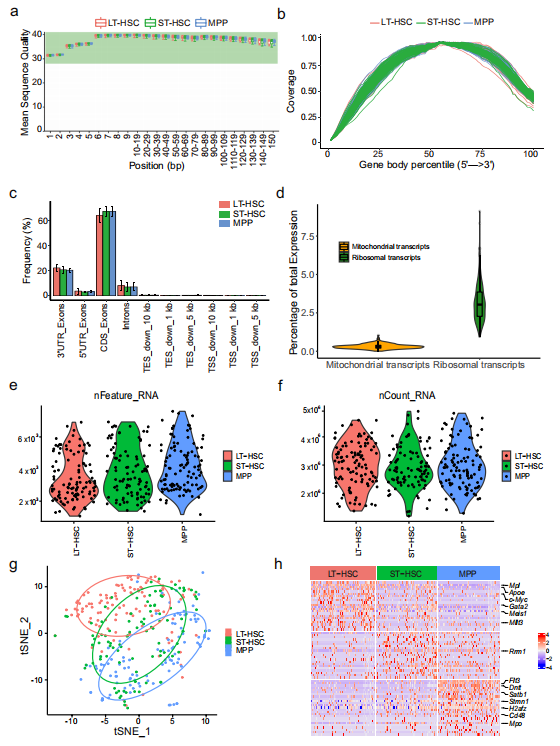
图2 单细胞短读长测序数据质控
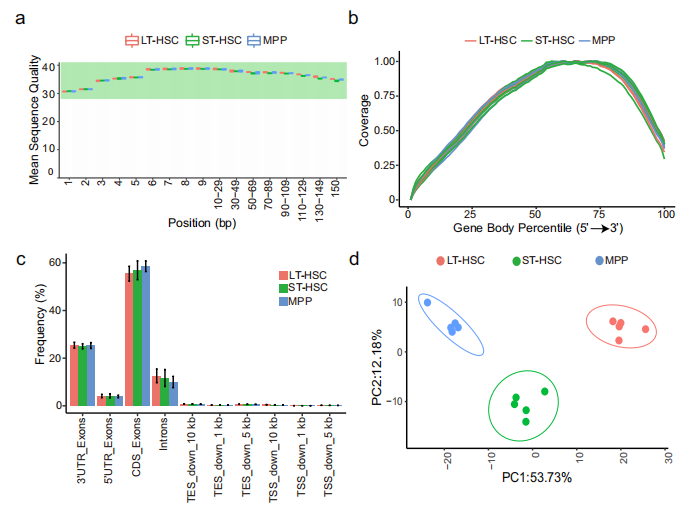
图3 批量细胞的短读长测序数据的质控
- 长读长测序数据的质控和一致性
纳米孔(ONT)测序数据的平均长度为1024 bp(图4a)。而PacBio测序数据的平均长度为946 bp(图4)。PacBio测序的质量得分高于纳米孔测序(图4b),平均值分别为47.57和10.53。接下来比较了长短读长测序在有无参考的情况下识别外显子和转录本的√准性。结果发现,无论有无参考,长读长测序都能提供相对完整的外显子链,包括转录水平上的新外显子(图4c,d),而当有参考时,短读长测序在识别外显子方面有着更高的√准性(图4d)。
为了评估重复之间的一致性,计算了短读长和长读长测序之间的基因定量的相关性。相关系数均在0.93以上(图4e),表明重复样本间具有较高的一致性。此外,PCA显示短读长和长读长测序数据按细胞类型进行了聚类(图4f)。结果表明,长读长测序数据质量高,生物重复一致性高。此外,长读长测序能够对新的外显子和转录本进行识别和定量。
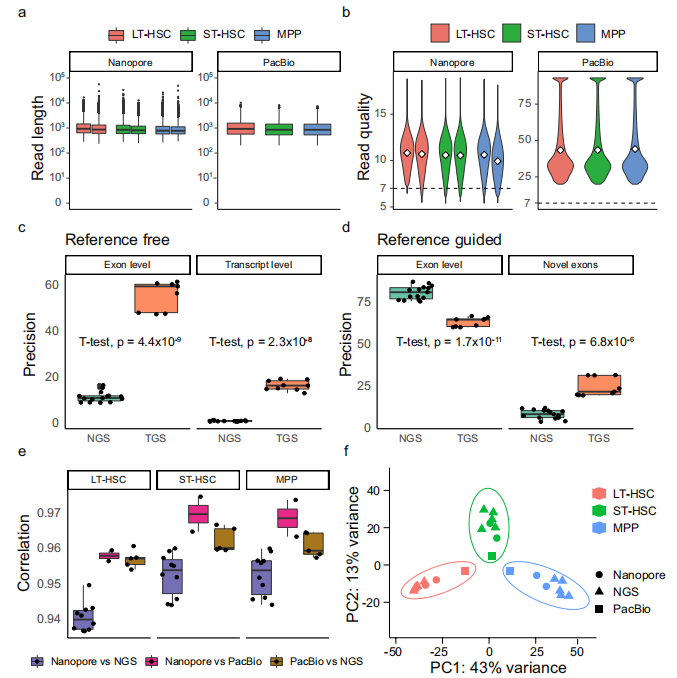
图4 批量细胞长读长测序数据的质控
- 整体可变剪接模式分析
为了研究使用长读长数据集的整体可变剪接模式,首先使用SUPPA2识别可变剪接事件和类型。有趣的是,在所有细胞类型中,常见的选择性剪接类型是保留内含子(RI),其次是外显子跳跃(SE)和可变3’或5’端剪接位点(图5a)。接下来发现超过21762个细胞型特异性的可变剪接事件(图5b)。SE是三个细胞类型中常见的可变剪接类型(图5c),其次是RI和可变3’或5’端剪接位点。这些结果表明,长读长测序有助于识别大量可能在造血过程中具有潜在功能的细胞特异性或共有的可变剪接事件。
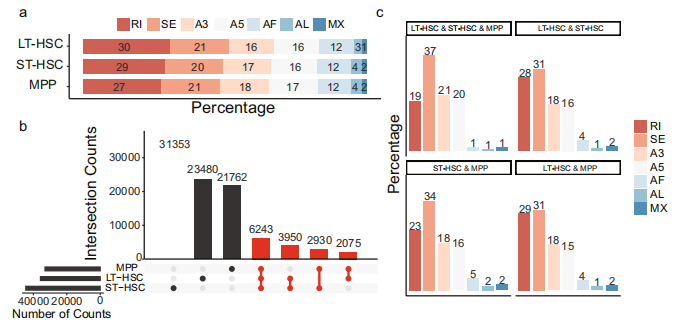
图5 批量细胞长读长测序数据的整体可变剪接分析
- 可变剪接异构体的鉴定和定量
为了进一步确认长读长在识别可变剪接异构体方面的优势,使用纳米孔和PacBio测序数据对三种细胞类型中已知的LT-HSC的标志物c-Myc和Gata2(图2h)的转录本进行了可视化。接下来筛选了所有映射到基因c-Myc和Gata2及其注释的转录本上的reads。发现2915个reads覆盖到了c-Myc,且LT-HSC的reads数*多(图6a)。在纳米孔和PacBio测序数据中可视化了全长转录本的reads,发现所有注释的亚型都能被鉴定识别(图6c)。在c-Myc第一个外显子中发现了一个5 ‘的可变剪接起始位点。使用短读长测序数据来定量该基因座的剪接百分比(PSI),发现较长的异构体在所有三种细胞类型中具有相似的 PSI(剪接百分比),ST-HSC中包含第一个外显子的较长异构体的reads占比比较高(图6 e)。对于Gata2来说,发现覆盖有595个reads,在LT-HSC中比其他两种细胞类型多了近20倍的reads数(图6b)。通过对比全长reads和注释的转录本,发现在LT-HSC中Ensembl的转录本中有一个未注释的内含子保留(图6d)。随后利用短读长测序数据验证验证了这种内含子保留并定量这个内含子的PSI值,发现该内含子在LT-HSC中PSI值*高(图6f)。接下来展示将长读长测序与单细胞RNA-seq结合的数据示例,从长读长测序数据中鉴定到了一个在Mpl中具有24 bp可变剪接区域的可5’剪接位点(A5)。基于Smart-seq2的测序数据发现这个剪接事件在不同细胞类型中是差异的,其PSI值从LT-HSC,ST-HSC到MPP是依次递减的(图6g-j)。使用Smart-seq2数据可以观察到单个细胞间的异质性(图6h),在造血过程中,所涉及的长和短剪接位点(SJ)是被显著下调的(图6i,j)。这些结果表明,整合短读长和长读长测序有助于识别差异表达的异构体。
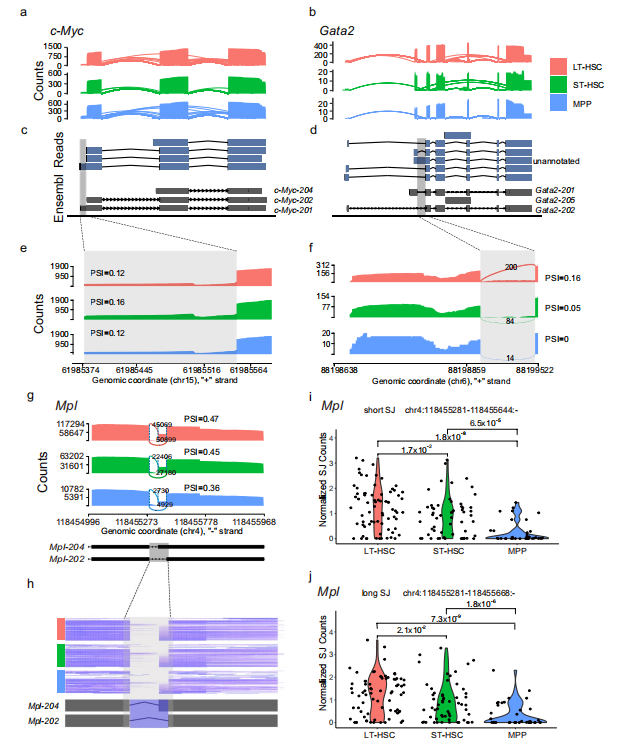
图6 长读长结合短读长数据识别定量可变剪接异构体
测序数据集说明
大批量短读长RNA-seq可在各种组织或细胞样本中用于√准性的定量基因表达和替代外显子使用。单细胞RNA测序在揭示细胞类型内基因表达的异质性方面是强而有力的。全长RNA-seq实验流程,如Smart-seq2,也可以检测可变剪接中的异质性。然而,使用短读长测序来组装转录本仍然很困难。
本文的测序数据集为揭示HSC群体中的转录本的多样性提供了独特的机会。通过整合短读长和长读长的批量测序数据集,可以更好地识别和定量(新型)可变剪接异构体。而scRNA-seq数据可以进一步提供有关这些转录本如何在不同HSC细胞类型中变化的信息。此外,该数据集可用于开发统计模型以重建异构体,并能够进一步在很大程度上研究未探索的转录后调控,例如单细胞水平的可变剪接和RNA编辑。
如果您对该研究思路感兴趣,点击下方按钮联系我们,我们将免费为您设计文章研究思路





 京公网安备 11011302003368号
京公网安备 11011302003368号