
Hi-C (High-through chromosome conformation capture) 是以整个细胞核为研究对象,利用高通量测序技术,结合生物信息分析方法,研究全基因组范围内整个染色质DNA在空间位置上的关系,获得高分辨率的染色质三维结构信息以及染色质调控元件相互作用图谱。Hi-C可以与RNA-seq、ChIP-seq、ATAC-seq和WGBS(全基因组甲基化测序技术)等数据进行联合分析,从基因调控网络和表观遗传网络来阐述生物体表型形成的相关机制。而其中ATAC-seq技术是继FAIRE-seq、MNase-seq、DNase-seq目前最火热的研究染色质开放性的新技术,自2013年后,文章的发表量逐年攀升。百迈客已做好承接各种组织类、细胞类样本的HiC互作和ATAC-seq的准备,成功案例和项目经验还缺您的一把火,年底大促销等您快快来咨询哦!
英文名称:Acute depletion of CTCF rewires genome-wide chromatin accessibility
杂志名称:Genome Biol.
影响因子:13.583
发表日期:2021年8月24日
摘要
CCCTC-结合因子(CTCF)是一种高度保守的含锌指转录因子,被称为“基因组编织大师”。它是研究最广泛的三维(3D)染色质结构调控因子。研究发现在CTCF急性衰竭细胞模型和其他基因敲除模型中,CTCF在全基因组TAD和TAD内部loop环的形成中是不可或缺的。为了更好地理解CTCF结合占用率如何促进转录调控,本文系统地进行了多组学研究,特别关注染色质可及性。通过系统整合了ATAC-seq、RNA-seq, WGBS, Hi-C, Cut&Run和CRISPR-Cas9基因筛选技术,以及深度蛋白质组学和磷酸化蛋白质组学分析,用以研究CTCF蛋白急性耗竭对细胞的影响。
材料方法
实验材料:人B细胞淋巴母细胞白血病(B-ALL)细胞系SEM(DSMZ),3个单细胞克隆(clones 27, clones35和clones42)分别用IAA处理24 h和48 h诱导CTCF退化。
ATAC-seq:有无IAA处理的三个克隆细胞样本,一式两份。
全基因组甲基化测序(WGBS):IAA处理24h或无IAA处理的clone27。
蛋白质组和磷酸化蛋白质组分析:CTCFAIDSEM细胞,分为4个组:no IAA,+IAA 12h,+IAA 23h和+IAA 48h,一式三份。
RNA-seq:有无IAA处理24h或48h的DSMZ。(前期研究)
Hi-C:有或者无IAA处理的clone27和clone35。(前期研究)
Cut&Run实验:有或者无IAA处理的clone35和clone42,选用CTCF抗体进行富集。(前期研究)
 一、急性CTCF耗竭改变了染色质的可及性
一、急性CTCF耗竭改变了染色质的可及性
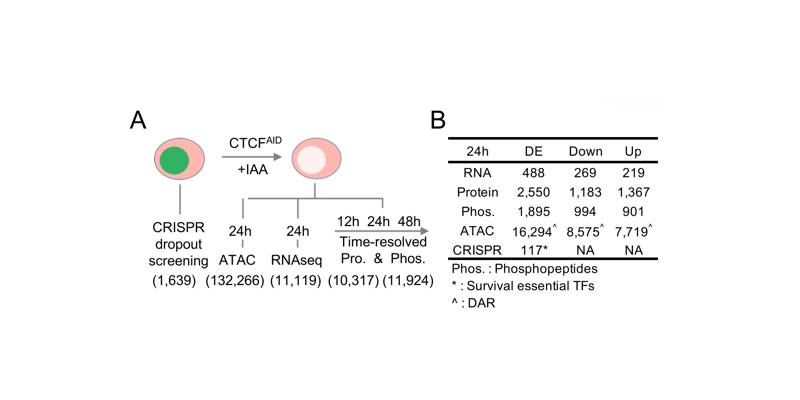
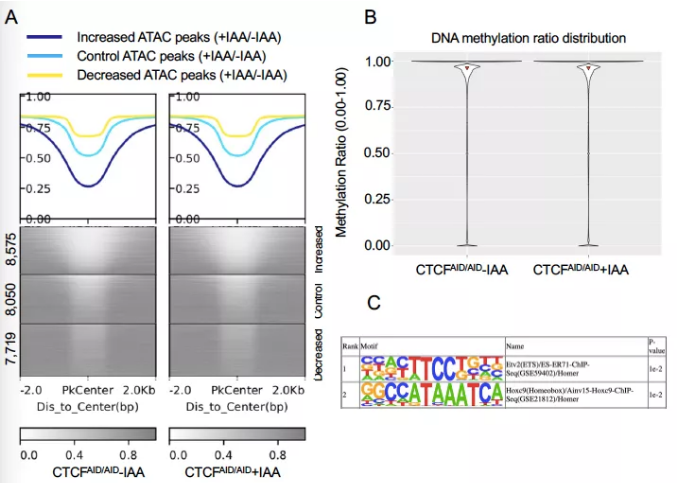
前期研究已经通过将双等位基因miniAID-mClover3标签导入人的内源性CTCF位点,并产生了三个CTCFAID细胞的克隆。在强力霉素和生长素(IAA)处理下,强制表达与Skp1/Culin/F-box (SCF)泛素连接酶组分连接的OsTIR1,快速降解CTCF融合蛋白(图1A)。当强力霉素和IAA完全从培养基中洗脱后,这种降解是可逆的。通过对三个单细胞来源的克隆进行IAA处理24小时的免疫印迹实验,证实了CTCF能够有效降解(图1B),类似于之前在48h处理条件下的研究结果。
为了研究CTCF耗竭对全基因组染色质可及性的影响,对有或无IAA处理的CTCFAID细胞进行了ATAC-seq,同时野生型SEM细胞和通过CRISPR敲除两个不相关靶点USF1和USF2的SEM细胞作为对照。总的来说,共发现了8876个显著降低的差异可及性区域(DARs), 8042个可及性显著增加的DARs。热图和峰值信号强度结果都证实了DARs具有高度重现性(图1C, D)。正如预期的那样,这些DARs在热图中的聚类效果显示出与CTCF耗竭相互一致的趋势,但在USF1/2敲除的SEM细胞中保持不变,表明这些DARs具有依赖CTCF的特征。
接下来分析这些DARs与它们最近的CTCF motif之间的物理距离。结果表明,可及性减弱的DARs更接近于最近的CTCF motifs。相比之下,可及性增强的DARs与CTCF motif的距离显著高于可及性减弱的DARs,但显著低于对照组(图1E)。
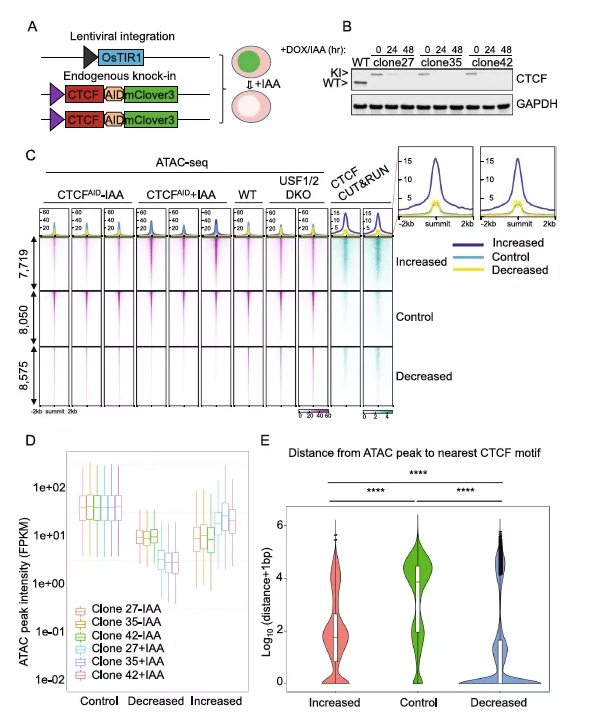
图1 急性CTCF耗竭会改变染色质的可及性
二、急性CTCF耗竭后染色质可及性特征发生变化
前面综合分析了在CTCF耗竭后,随着染色质可及性的改变,转录因子的占用情况。接下来通过查看motif数据库TRANSFAC中所有带注释的TF motif,在三类区域中计算他们的富集频率:减弱的DARs、增强的DARs和对照区域。发现在减弱的DARs中最富集的TFs是CTCF和黏连蛋白(SMC3和RAD21)(图2A)。Tn5插入位点的foot-printing分析证实,它们的motifs在motif中心受到保护(图2C)。这些结果表明,减弱的DARs反映了CTCF耗竭产生的影响。
增强的DARs也富集到了CTCF motif,与之前的CTCF-motif距离分析一致(图1E)。然而,最富集的TF不是CTCF motif。相反,有许多是与活性转录相关的一般转录因子(GTFs)(图2B, D)。这些数据说明DARs的调控作用很可能和CTCF的抑制功能相关。
虽然CTCF和黏连蛋白的motifs在增强和减弱的DARs中都富集到了,但它们在增强的DARs中的foot-printing分析表现出不同的模式特征。与减弱的DARs中Tn5保护的motif中心相比,这些motif周围的近端侧翼区域更受保护,这与活性启动子和增强子相关的串联CTCF motif(2xCTSes)一致。结果发现,8042个区域中有1244个(15.4%)的DARs与2xCTSes重叠,比对照区域和减弱DARs区域更富集。而这些2xCTSes被认为调节染色质loop环,观察到的DARs可能与染色质loop环直接相关。
接下来,将这些loop环分成三组,并用不同的标准绘制它们的正常染色质接触数。与增强的DARs重叠的loop环展现出更多的染色质内接触,而与减弱的DARs重叠的loop环展现出更少的染色质内接触。三组的染色质内接触均在CTCF耗竭后减少。然而,重叠于减弱的DARs的loop环减少的触点显著多于重叠于对照NFRs区域和增强的DARs的loop环(图2E)。总的来说,loop环的形成可能只反映CTCF的结合状态,而不是直接调控染色质可及性。然而,较弱的远端环似乎更容易失去CTCF。
最后尝试探索这些DARs是否与TAD边界有关,发现对照的ATAC-seq峰和减弱的DARs到TAD边界的距离分布相似,而增强的DARs总体上出现在远离TAD边界的地方(图2F)。已知TAD边界在CTCF结合位点和转录活性基因(包括管家基因)中富集。CTCF占据的物理位置似乎与其转录调控密切相关。
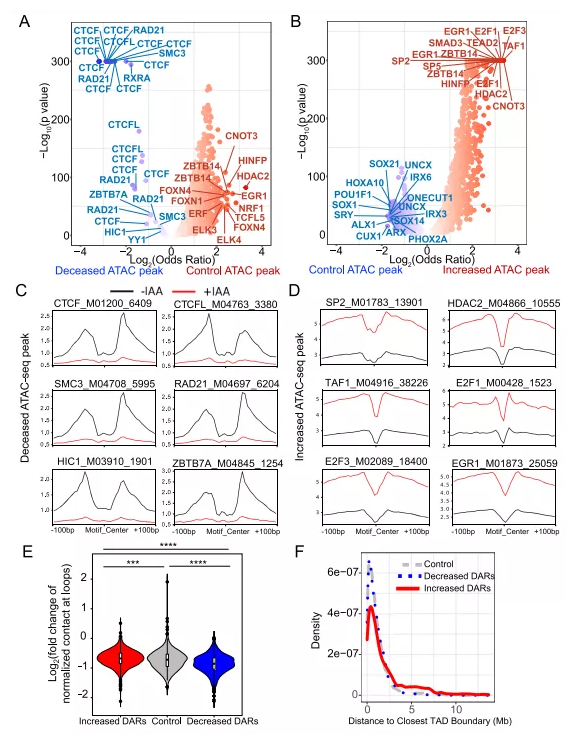 图2 急性CTCF耗竭后染色质可及性的特征变化
图2 急性CTCF耗竭后染色质可及性的特征变化
本文假设急性CTCF耗尽的细胞模型最适合于确定全基因组DNA甲基化的即时反应。
令人惊讶的是,当用WGBS生成DNA甲基化谱时,并没有观察到急性CTCF耗尽后全基因组DNA甲基化的变化。与ATAC-seq和CTCF Cut&Run的分析结果不同,DARs周围的DNA甲基化水平在对照组和CTCF耗竭组之间没有发现有差异性(图S9A)。接下来分析差异甲基化区域(DMRs),发现只有49个显著差异区域(图S9B)。进一步检查这些增强的DMRs富集的motif,并没有发现CTCF或黏连蛋白(图S9C),表明这些DMRs与CTCF占用没有直接关联。总之,研究结果表明,急性CTCF耗竭不会影响SEM细胞中全基因组DNA甲基化。
 图S9 急性CTCF耗竭不影响全基因组DNA甲基化
图S9 急性CTCF耗竭不影响全基因组DNA甲基化
三、依赖CTCF的染色质可及性通过启动子或增强子-启动子loop环调节基因表达
虽然CTCF在某些位点的基因调控中不可或缺,如H19-IGF2、β-血红蛋白、原钙粘蛋白簇和TP53等,但目前尚不清楚这种转录调控是否由CTCF直接作用,或者染色质可及性是否也发挥了作用。火山图显示增强的DARs中基因启动子数比减弱的DARs中的更多(图3A)。接下来计算DARs中的基因数,发现这些基因在IAA处理后的细胞的RNA-seq数据中也表现转录差异。更多减弱的DARs往往与下调的基因相关,而更多的增强的DARs通常与基因表达增多相关。
对于表现出一致变化的基因,进一步检查它们启动子的ATAC-seq信号,并确认模式与预期一致(图3B)。还使用基因表达水平和ATAC-seq信号z-score制作了热图,确认了可重复的模式(图3C)。基于排名最高的基因集进行了基因集富集分析(GSEA),结果表明减弱的DARs与基因下调相关,而增强的DARs与基因上调相关。因此得出结论,与DARs相关的转录变化特征直接响应CTCF的急性耗竭。
CTCF基因启动子的染色质可及性在CTCF耗竭时也有所增加(图3D),这一点通过定量PCR (Q-PCR)进一步得到验证(图3E)。这些数据表明CTCF可以抑制自身以保持最*表达水平。对于CTCF耗竭后被下调的基因,如MYC,并没有在启动子区域检测到有统计学意义的染色质可及性变化。因此,当启动子区域保持开放时,远端增强子区域的染色质景观变得更难以接近,再加上CTCF耗竭,可能在控制能调控MYC转录的远端增强子-启动子loop环的形成中发挥作用。
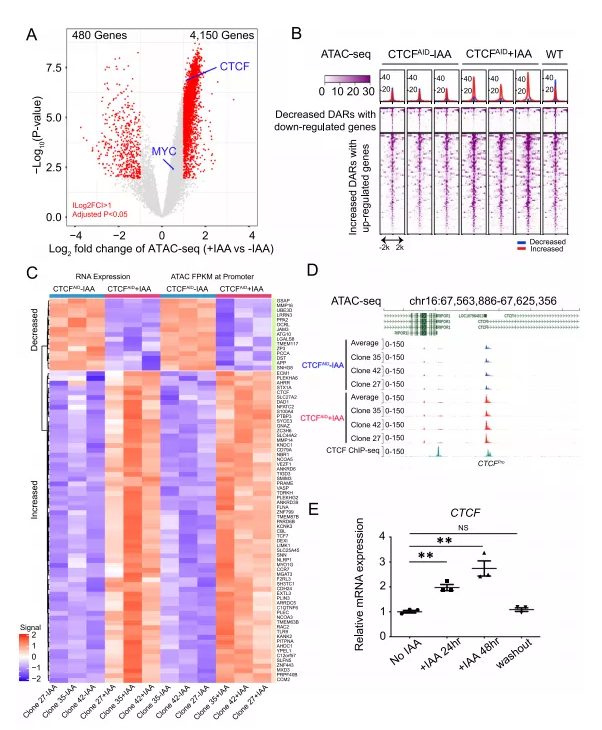 图3 启动子或增强子-启动子环调节基因表达
图3 启动子或增强子-启动子环调节基因表达
四、综合分析假定的绝缘子CTCF结合位点
本文通过综合分析建立了一个框架来识别假定的CTCF介导的绝缘子元件。将ATAC-seq结果中的3490个峰和有CTCF结合的峰取交集,共有716个增强的ATAC-seq峰。接下来,将它们与RNA-seq结果中上调的基因进行比对,判断TSSs是否位于距离DARs区域2-50 kb的范围。综上所述,有67个基因符合这些标准(图(Fig 4A)。这67个基因中有20个基因的附近有染色质loop环。例如,在BLCAP基因上游约7 kb处观察到一个假定的抑制性CTCF结合峰,该峰位于Hi-C数据所示的染色质绝缘loop环中(图4B )。在未经过IAA处理的对照CTCFAID细胞中,CTCF与该motif结合导致染色质可及性受到抑制,这在ATAC-seq数据中信号的缺失很明显。然而,在急性CTCF耗竭时,ATAC-seq峰值信号和BLCAP mRNA表达明显增加(图4C)。
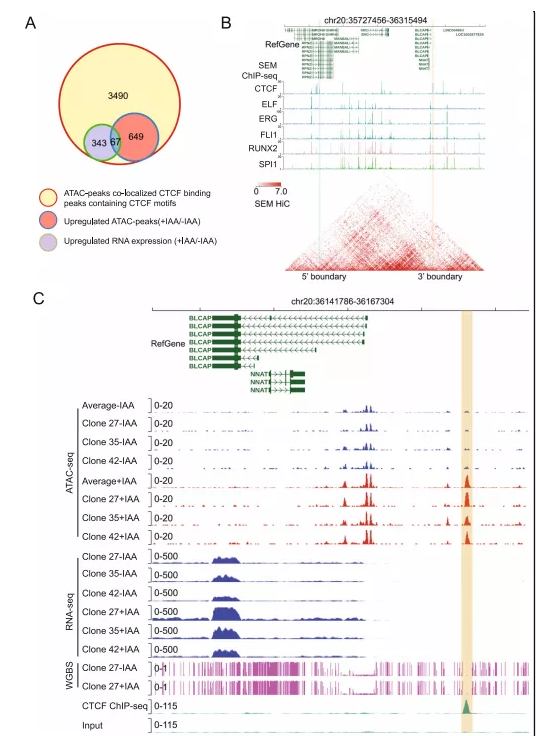 图 4 综合分析假定的绝缘子CTCF结合位点
图 4 综合分析假定的绝缘子CTCF结合位点
五、CTCF抑制BLCAP表达的功能验证
为了进一步验证预测的假定绝缘子的作用,将慢病毒表达的引导RNA感染表达Cas9的SEM细胞中,然后进行抗生素选择。Sanger基因组测序(Inference of CRISPR edit, ICE)在目标人群中检测到约61%的总indel频率(图5A),与非靶向导向对照组(sgNT)相比,导致BLCAP mRNA表达显著增加(图5B)。用IAA处理CTCFAID细胞24和48小时,然后洗脱IAA进行CTCF修复。通过RNA-seq分析和Q-PCR验证,验证急性CTCF蛋白耗竭时BLCAP mRNA的表达(图5C)。不出所料,生长素处理24或48 h后CTCF蛋白急性耗竭,BLCAP表达水平显著升高。更重要的是,洗脱生长素后,其表达水平恢复到亲本细胞的水平(图5D)。综上所述,这些数据有力地支持了CTCF在BLCAP调控区域的占用充当了控制BLCAP表达的功能绝缘体。
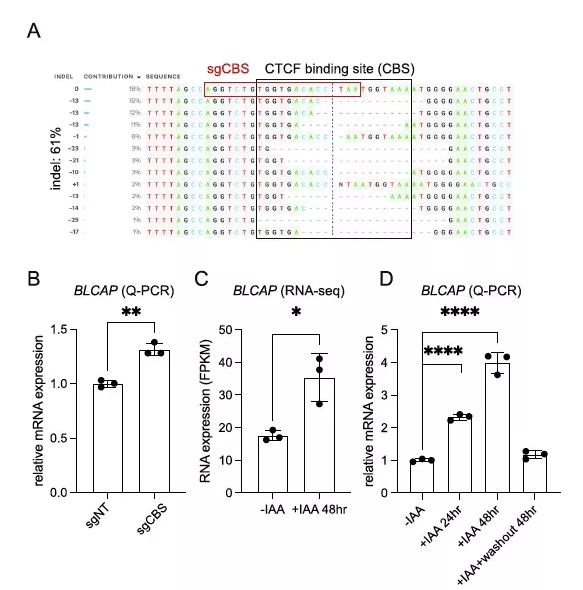
图 5 CTCF在抑制BLCAP表达方面的作用的功能验证
六、多组学联合揭示CTCF共同调控因子
为了进一步研究急性CTCF耗竭对基因表达的影响,本文系统探索了CTCF介导的下游蛋白组和磷酸化蛋白组水平的基因表达,并将其与ATAC-seq和RNA-seq数据联合分析(图6A)。总的来说,将24小时治疗组与无IAA治疗组比较,确定了2550个差异表达蛋白质和1895个差异表达磷酸肽(图6B)。虽然观察到整体蛋白质组和转录组之间存在合理的相关性(图6C),但只有488个DE mRNA。与免疫印迹和Q-PCR结果一致,基于质谱(MS)的蛋白质组学和RNA-seq分析证实,急性CTCF耗竭后,在蛋白水平上CTCF表达显著减少,在mRNA水平上表达增加。这些数据表明,尽管mRNA水平的变化并不明显,急性CTCF耗竭诱导了下游响应的大量中断。
本文开发了一种多组学联合的方法来定义CTCF共调控转录因子,总共鉴定了40个CTCF共同调控因子,这些TF在急性CTCF耗竭时在mRNA和/或蛋白质水平上显著影响其下游靶基因的表达(图6D)。正如预期的那样,这40个CTCF共调控因子中的大多数也在CTCF介导的DARs中有共同定位,证实了它们与CTCF有着潜在的直接共同调控作用。
此外,我们还研究了CTCF的共调控模式,并选择了候选的TF包括 ZBTB7A和YY1,同时DUX作为阴性对照来观测。与对照和增强的DARs相比,这两个motif的绝大多数距离减弱的DARs更近,这表明CTCF的耗竭可能会影响相邻的开放染色质可及性,导致与其他TFs结合的缺失(图6E,F)。而DUX4和CTCF motif距离分布均匀(图6G)。总之,本文系统地揭示并验证了CTCF介导和招募的主要共调控因子,通过编织和改变染色质可及性来实现下游转录调控,并证明了多组学分析方法是强大的,可以识别不具有明显表达变化的隐藏主调控因子。
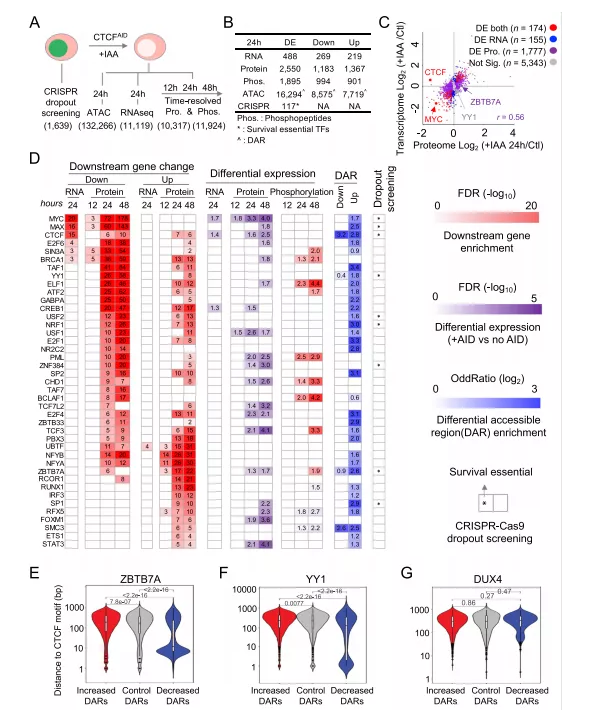 图6 多组学分析CTCF调节下游基因表达的主共调控因子
图6 多组学分析CTCF调节下游基因表达的主共调控因子
讨论
使用急性CTCF退化系统和丰富的可用数据集提供了直接证据,表明CTCF调控染色质可及性,但不调控DNA甲基化。CTCF可能在串联CTCF结合位点维持染色质可及性,从而招募CTCFL到附近的基因并启动转录。虽然CTCF的急性耗竭会深刻地干扰整个染色质相互作用和可及性,但转录水平通常不会有显著改变。这些发现表明在CTCF急性耗竭后蛋白质翻译和翻译后修饰过程中发生了潜在的全局变化。总之,CTCF的急性耗竭改变了染色质相互作用和可及性,需要进一步的研究来更好地理解CTCF的耗竭如何导致蛋白质和翻译后修饰的巨大变化。
如果您对HiC&ATAC-seq测序技术感兴趣,欢迎点击下方按钮联系我们,我们将免费为您设计文章思路方案。




 京公网安备 11011302003368号
京公网安备 11011302003368号