
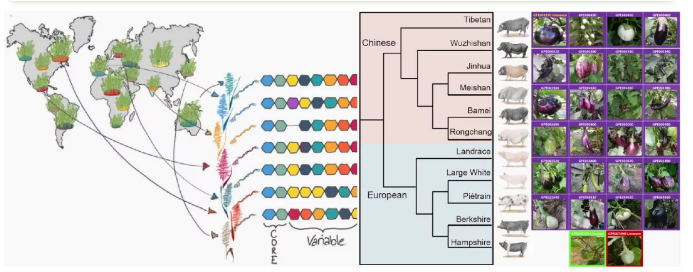
物种中不同品系表型差异示意图
(Jayakodi M et al., DNA Research; Tian X et al.Sci China Life Sci; Barchi L et al.,Plant J)
因此对于一个物种来说,如果只使用单一的参考基因组进行遗传驯化变异的研究,可能会丢失掉大量有意义的基因信息。为了解决这些问题并充分理解关键物种中重要性状形成机理,为育种奠定重要的研究基础,动植物泛基因组研究应运而生。
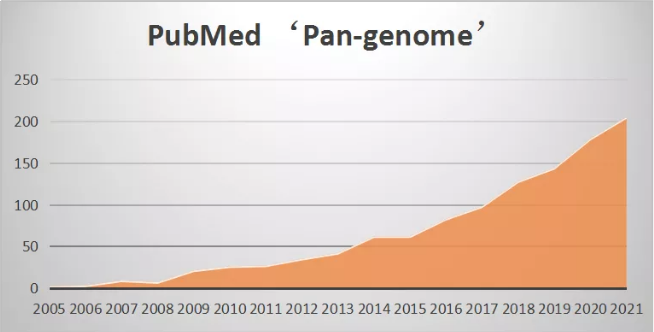
泛基因组近年发展趋势(基于NCBI PubMed搜索pan-genome)
泛基因组分析有助于理解物种的特征,同时泛基因组图谱提供的复杂基因组变异,有助于解析作物表型和农艺性状的多样性。选择不同亚种材料进行泛基因组测序,可以研究物种的起源及演化等重要生物学问题;选择野生种和栽培种等不同特性的种质资源进行泛基因组测序,可以发掘重要性状相关的基因资源,为科学育种提供指导;选择不同生态地理类型的种质资源进行泛基因组测序,可以开展物种的适应性进化、外来物种入侵性等热门科学问题。
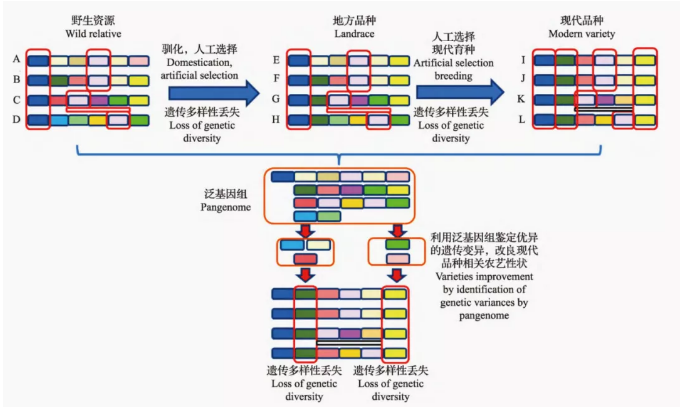
泛基因组在遗传变异发掘和植物分子育种中应用[1]
其实说了很多泛基因组的概念与意义,那么泛基因组到底能分析哪些内容呢?接下来容小编一一道来~

泛基因组构建的开放性检测
广义的泛基因组是指获取该物种全部遗传信息,构建一个非冗余的集合体。由于个体间的差异,在构建泛基因组的时候会在参考基因组的基础上,不断引入新的特异性序列。因此,统计结果可发现整个泛基因组的大小随着个体数量的增加而增加,核心基因组(几乎存在与全部个体基因组中)的大小随着个体数量的增加而减小(如图1)。
由于物种受到生存环境与生态位差异、有效群体规模、多态性水平差异等因素的影响,泛基因组呈现开放型和闭合型两种模式。对于闭合型的泛基因组,核心基因组和整个泛基因组含量会随着个体增加很快到达平台期。而开放型泛基因组的物种具有更丰富的遗传资源库、更强的多态性与环境适应性,因此想达到平台期通常需要更多的个体加入,才能通过有限的个体充分展示物种内全部遗传信息。从而,通过泛基因组与核心基因组的比例统计,可以了解所选材料构建的泛基因组是否具有一定的代表性。
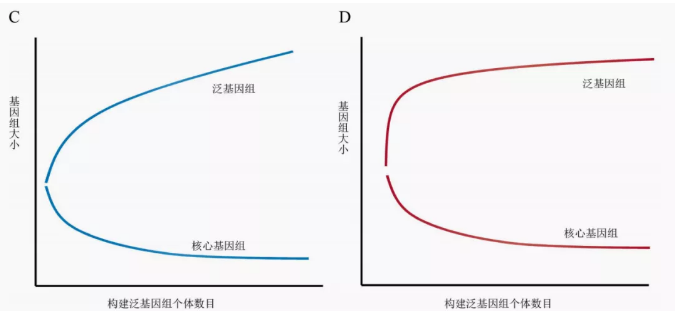
图1开放型(左)与闭合型(右)泛基因组[2]
泛基因组核心与非核心基因分析
泛基因组分析通过对每个个体中共有情况进行基因集聚类,通常分为以下三种:
核心基因(core gene):在所有动植物品系或者菌株中都存在的基因;非核心基因(dispensable gene):在1个以及1个以上的动植物品系或者菌株中存在的基因。特有基因(Private):仅在一个品系中存在的基因。
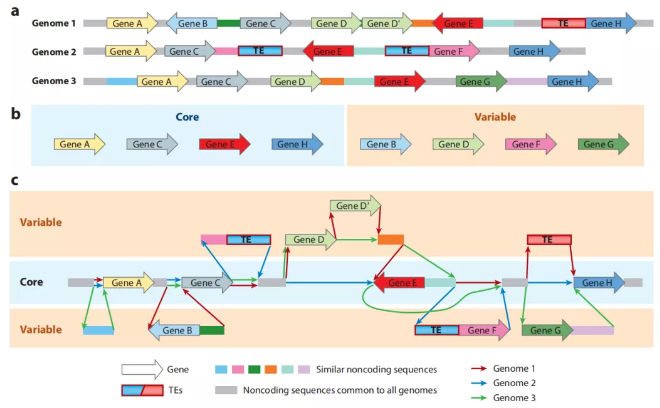
图2 核心基因与非核心基因示意图[3]
随着基因组数量的增加,核心基因的识别将严重受到denovo中组装错误的影响。为了纠正这种错误,一些科研工作者将泛基因组进一步细分:在所有基因组的为核心基因(core),在几乎所有品系中发现的为soft-core,在比soft-core少但不止几个品系的品系中发现的shell,以及在只有几个品系中发现的为cloud。具体的分类比例与样品数量间的关系并不固定,目的均是为了对基因进行分组,以便进行可靠的比较(图3)。而这些分组的基因频率呈现不对称的首尾高中间低的分布形式,充分展示了不同类别基因型的占比情况。
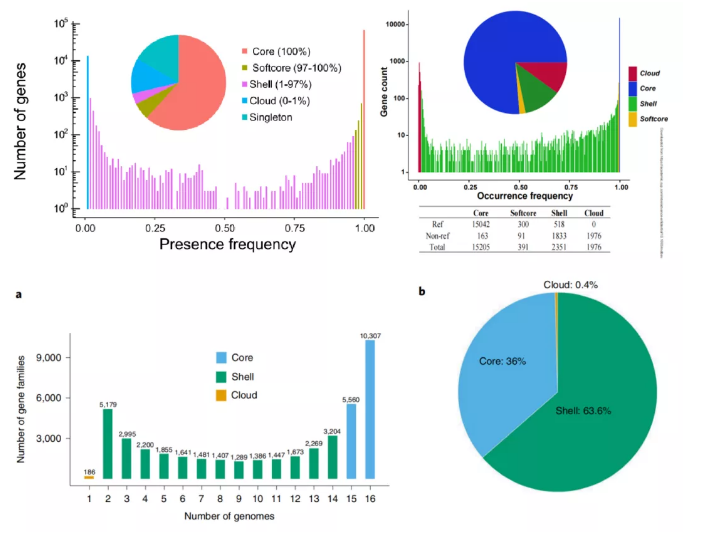
图3 棉花(上左)、鸡(上右)与高粱(下)泛基因组成分
(Li J et al.Genome Biol.2021;Wang K et al.,Mol Biol Evol.2021;Tao Y et al.,Nat Plants.2021)
除此之外,根据core、dispensable等各组分聚类结果构建基因家族的存在与缺失分布热图,进一步了解不同材料中的差异情况。同时可结合不同来源or不同表型等特征充分挖掘其基因型的多态性。

图4 鸡泛基因组中每个个体基因家族的存在缺失分布图
(Wang K et al.,Mol Biol Evol.2021)
从进化的角度来讲,pan-genome中鉴定出的核心基因(core genes)可能是执行关键功能的基因,其在一个物种中倾向于一些保守的基因。相反,非核心基因促进了物种的多样性,使其能够适应各种环境条件。核心基因通常富集到控制着生命体基本生成代谢的基本细胞功能,而非核心基因则富集到与环境和防御反应,受体和抗氧化剂活性,基因调控以及信号转导等相关的功能。通过功能注释可了解到核心、可变与特有基因家族的功能情况,从而找寻物种特异的功能与性状形成相关通路(图5)。
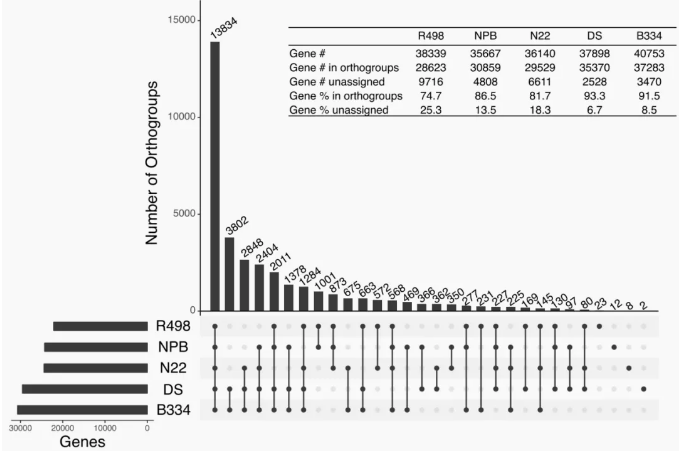
图5 水稻共有和特有基因家族分析
(Choi J Y et al., Genome Biology.2020)
例如,地中海贻贝泛基因组研究中发现,可变基因显著富集在与环境适应性相关的通路中(如细胞凋亡、免疫信号传导等),增强了地中海贻贝的环境适应性(图6)。
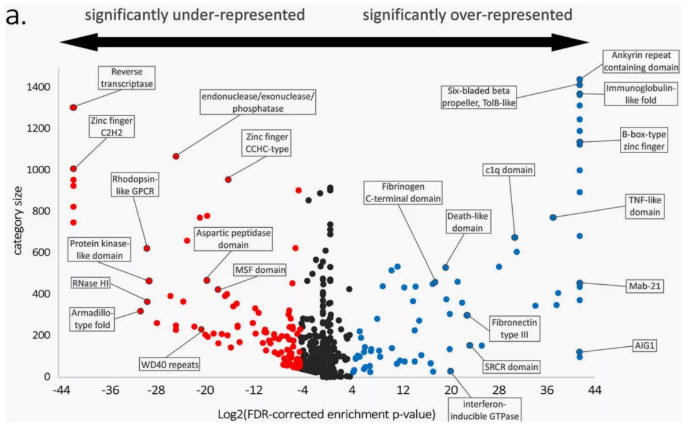
图6 贻贝泛基因组表达情况
(Gerdol M et al., Genome Biology.2020)
众多研究表明,非核心基因比核心基因的可变性更强;与核心基因集相比,非核心基因集中的非同义突变比同义突变的比率要高。因此通过同义与非同义突变(ka/ks)可以用来检测非核心基因集中基因是否受到选择作用。
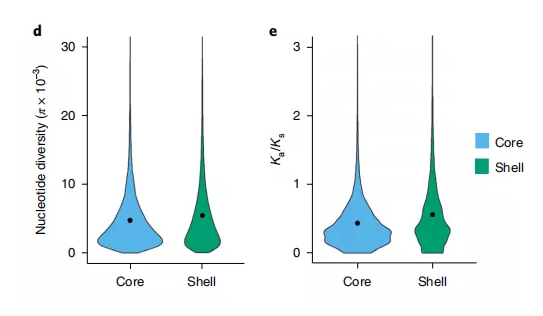
图7 高粱泛基因组Ka/Ks占比情况
(Tao Y et al.,Nat Plants.2021)
泛基因组变异分析
结构变异是泛基因组的重点研究对象,这些变异信息为物种的人为选择及自然选择提供原始的材料。了解变异产生的过程,探究不同材料的变异程度并利用有利的变异对维持并提高物种生产力至关重要,有助于解析物种表型和农艺性状的多样性。泛基因组研究,为更好地了解SV在动植物驯化中的作用提供了基础。主要研究的变异类型为:SNP(Single nucleotide polymorphism),即单核苷酸多态性变异,主要指在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性,这种变异可由单个碱基的转换(transition)或颠换(transversion)所引起。InDel(Insertion-Deletion),指的是在基因组的某个位置上所发生的小片段序列的插入或者删除,其长度通常在在 2 bp-50 bp 之间。SV(Structure Variantions),即基因组结构性变异,通常指的是基因组上大长度的序列变化和位置关系变化,有丰富的变异类型,包括长度在50bp以上的长片段染色体倒位、染色体易位、重复、存在/缺失(PAV)、拷贝数变异(CNV)。
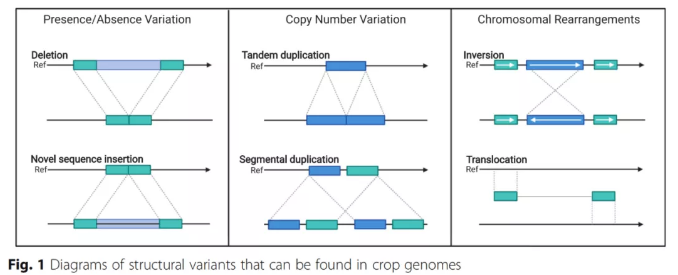
图8 SV变异类型[4]
例如高粱泛基因组研究发现,相对于参考基因组,其它高粱个体基因组中有429~1118个基因存在拷贝数变异(CNV)。功能注释显示这些CNV基因丰富了防御反应、信号转导和细胞蛋白分解代谢等生物学过程。并且不同高粱个体基因组上存在大量的存在缺失变异(PAV),有2514个基因受PAV严重影响,这些基因富集于与生物和非生物胁迫相关的生物过程,而抗病基因在这组基因中显著富集。进一步研究发现Yellow seed1、SbRc的PAV影响到高粱籽粒颜色变化(图9)
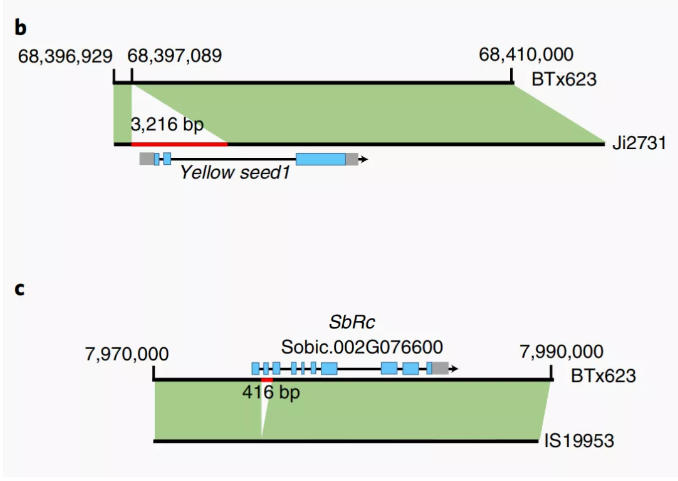
图9 Yellow seed1 基因和sbRc基因的序列差异
(Tao Y et al.,Nat Plants.2021)
除此之外,在其他物种中同样也发现大量结构变异导致性状差异。在水稻中,主要基因的presence/absence变异赋予其对淹没这一环境的耐受性,如Sub1A基因的PAVs变异(Xu et al., 2006),两个乙烯反应因子基因,SNORKEL1和SNORKEL2,进一步引发了深水水稻与非深水水稻对水深的不同反应(Hattori et al., 2009)。水稻泛基因组研究发现SV热点区域与稻瘟病抗性QTLs重叠/邻接,OsGLP2-1的存在可能保证种子的适当休眠,有助于其适应环境。
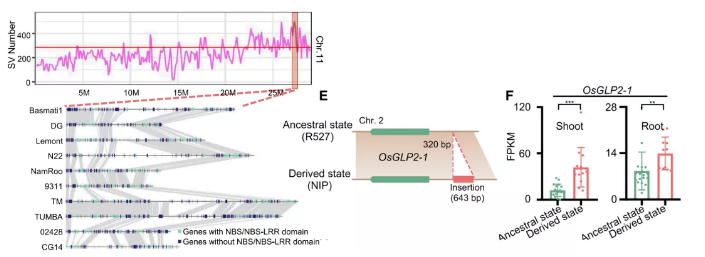
图10 水稻SV热点区域与种子休眠相关基因插入
(Qin P et al., Cell.2021)
众多泛基因组SV分析结果中我们不难发现,SV在农作物重要农艺性状的遗传控制中起着关键作用。基因组的结构变异可以导致物种个体间基因结构、表达特性、基因剂量等发生实质性地变化,并对动植物农艺性状形成了深刻的影响。
图形泛基因组构建
图形泛基因组是指以参考基因组为框架,将其他个体材料基因组序列与之比对,存在差异的地方构成不同的分支,并随着新序列的加入不断扩展变化,最终构建出一个包含全部变异信息的复杂图形结构(图11)。图形泛基因组可以更好的展示每部分序列在泛基因组中的位置关系,使构建一个包含物种内全部遗传信息的泛基因组成为可能。图形泛基因组已经在大豆、水稻、牛、高粱等物种有了初步构建与应用,为基因组学研究提供了一个重要的推动力。
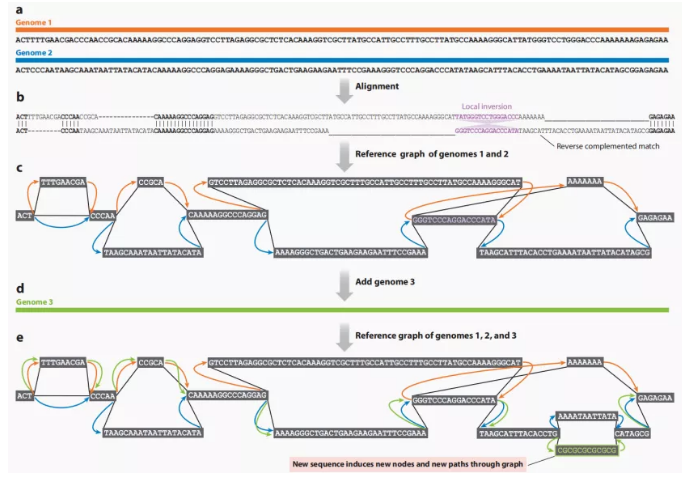 图11 图形泛基因组构建过程[3]
图11 图形泛基因组构建过程[3]
转座子分析
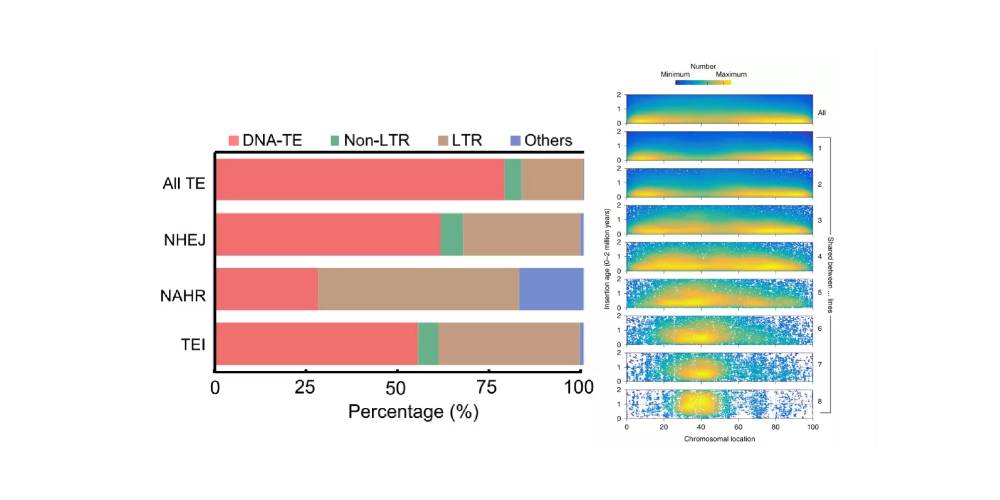
TEs转座因子(Transposable element,TE):又称之为“跳跃基因” “可移动的遗传因子”,是基因组中最活跃的部分,TE的活性被认为是不同物种之间基因组大小变化的重要因素。SVs可以通过TEs转座子的移动而形成(主要位于非核心基因区),在很多物种中已经发现PAVs富集在转座子TEs区,TE活性可以介导大规模的染色体重排。随着时间推移,转座子两端的LTR由于碱基突变逐渐产生了差异。通过这种与时间长短有关的序列差异,利用LTR之间的分化距离计算转座子插入至基因组的时间,进而可以评估不同品种的遗传进化关系。
水稻中TEs,尤其是LTRs促进非等位同源重组(Qin P et al.,Cell.2021);玉米泛基因组中随着个体的增加,LTR插入呈现向中心富集,并与插入时间的增加相关。
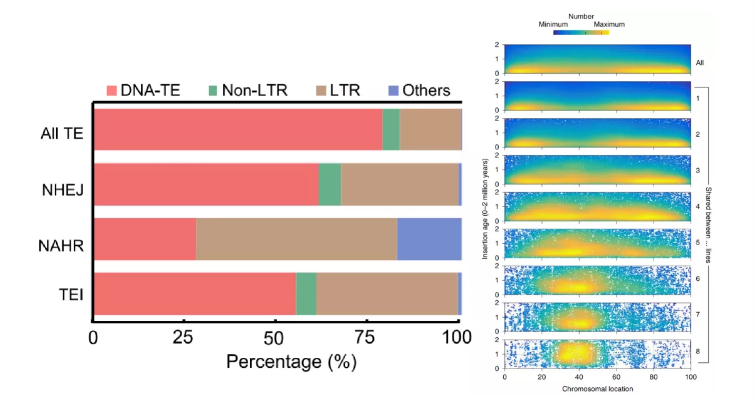
图12 水稻中TE类别比例与玉米LTR插入时间与分布
(Qin P et al., Cell.2021;Haberer G et al., Nat Genet.2020)
群体变异挖掘
由于泛基因组保留了群体完整的基因组多样性,可鉴定到单一参考基因组无法鉴定的基因组变异。泛基因组结合GWAS、QTL等数据,可进一步提高变异与性状关联的精度,捕获到更加完整的遗传变异信息,植物遗传学家和育种者提供全面的基因组资源,对于物种育种和研究具有重要的意义。例如玉米泛基因组研究发现,在检测到的GWAS信号中,关联信号中SV相关的信号有93.05%与SNPs识别的信号重叠,SV特有信号最显著的是关联到10号染色体上的北叶枯病QTL。该SV位于编码thylakoid lumenal protein的基因内;这些蛋白质可以通过调节病毒感染期间的细胞死亡与植物免疫关联。
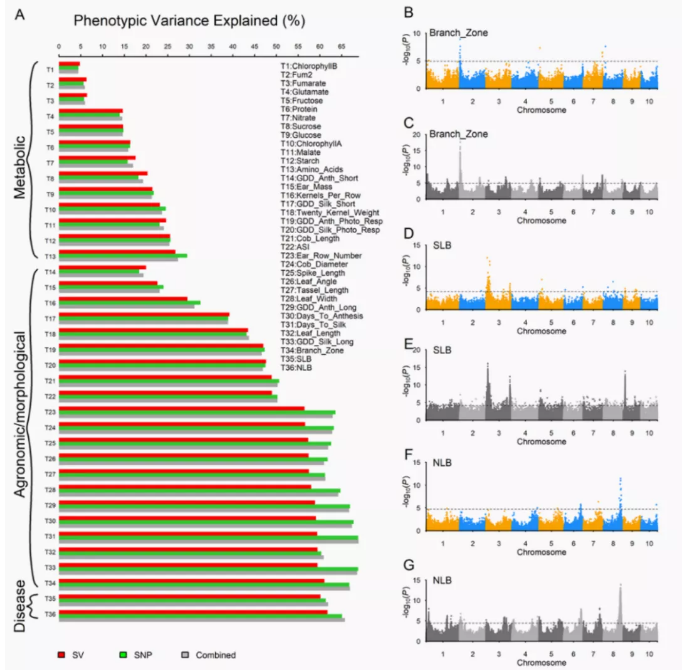
图13 玉米SV 与SNP GWAS(Hufford MB et al., Science.2021)
前期泛基因组系列文章已举非常多的案例,这里不再反复阐述~(点击回顾:泛基因组(Pan-genome)研究思路与应用—作物篇)
泛基因组研究展现了作物多样性和改良的前景,总的来说其研究内容主要集中在不同生态型、表型等品系材料间共同与差异的基因与功能研究。重要的是这些变异能被挖掘并为育种提供重要的理论基础。
而更重要的是,以上内容百迈客均可进行分析,图片还老好看了;更更重要的是,百迈客可得到的结果还远不止以上这些
如果您对我们的服务产品感兴趣,欢迎点击下方按钮联系我们,我们将免费为您设计文章思路方案。

参考文献:
[1]赵均良,张少红,刘斌.泛基因组及其在植物功能基因组学研究中的应用[J].植物遗传资源学报,2021.
[2]边培培,张禹,姜雨.泛基因组:高质量参考基因组的新标准[J].遗传,2021.
[3]Lei L , Goltsman E , Goodstein D , et al. Plant Pan-Genomics Comes of Age[J]. Annual Review of Plant Biology, 2021.
[4]Coletta R D, Qiu Y, Ou S, et al. How the pan-genome is changing crop genomics and improvement[J]. Genome Biology, 2021.



 京公网安备 11011302003368号
京公网安备 11011302003368号