
ATAC-seq技术是继FAIRE-seq、MNase-seq、DNase-seq目前最火热的研究染色质开放性的新技术,自2013年后,文章的发表量逐年攀升。百迈客已成功开展各种组织类,细胞类样本的ATAC-seq,项目经验积分满满哦!基于ATAC-seq数据怎么开展数据分析,这里有篇简单易学的套路文章,一起看看吧?
英文名称:The landscape of accessible chromatin in quiescent cardiac fibroblasts and cardiac fibroblasts activated after myocardial infarction
杂志名称:Epigenetics
影响因子:4.521
发表日期:2021年10月25日
摘要
心肌梗死(MI)后,心肌细胞大量死亡,导致心脏成纤维细胞增殖和分化,促进梗死心肌细胞外基质重塑。最近发现心脏成纤维细胞可以进一步分化为基质纤维细胞,这是一种新发现的心脏成纤维细胞的分化状态。不同状态的心脏成纤维细胞具有与其功能密切相关的不同基因的表达谱。然而,在这些激活和分化事件发生期间引起基因表达变化的机制仍不清楚。
材料方法
RNA-seq:新鲜分选的来自小鼠未受伤心肌(静止的心脏成纤维细胞)和心肌梗死后第 3 天(早期增殖成肌纤维细胞)、第 7 天(成熟成肌纤维细胞)、第 2 周(过渡基质成纤维细胞)和第 4 周(基质成纤维细胞)梗死区的心脏成纤维细胞,每组设置两个重复
ATAC-seq:新鲜分选的来自小鼠未受伤心肌和心肌梗死后第3天、第7天、第2周、第4周梗死区的心脏成纤维细胞,每组设置两个重复
实验方法:共聚焦显微镜成像,流式细胞分选
结果
一、 不同分化状态下心脏成纤维细胞的转录组分析
本文采用他莫昔芬对小鼠进行处理,并手术诱导心肌梗死(MI)(图 1a,b)。未受伤心肌和梗死区的共聚焦成像显示MI后心脏成纤维细胞数量显著增加,心脏成纤维细胞也变大了(图1c,d))。
对未受伤心肌和梗死区分选出来的心脏成纤维细胞进行RNA-seq分析(图2),发现不同状态的心脏成纤维细胞的转录组数据有明显不同。与来自未受伤心肌的心脏成纤维细胞相比,总计5000个左右的差异基因中有3829个基因在MI后的心脏成纤维细胞差异性表达。差异基因热图结果表明增殖相关的基因的表达在MI后第3天达到峰值,应激纤维基因、心肌ECM基因和细胞迁移基因在MI后第3天和第7天表达量最高。
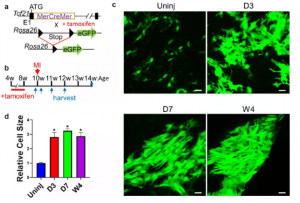
图1 MI后心脏成纤维细胞的谱系追踪
 图2 不同分化状态下心脏成纤维细胞的RNA-seq分析
图2 不同分化状态下心脏成纤维细胞的RNA-seq分析
二、 不同分化状态下心脏成纤维细胞的染色质图谱
对未受伤心肌和梗塞区的心脏成纤维细胞进行了ATAC-seq分析(图3),来研究在MI后心脏成纤维细胞的激活和分化过程中染色质的可及性。结果发现ATAC-seq峰在转录起始位点 (TSS) 和转录终止位点 (TES) 区域以及外显子区域高度富集,而且ATAC-seq峰在可能包含许多启动子的CpG岛上高度富集,表明DNA甲基化可能在心脏成纤维细胞的激活和分化过程中的染色质重塑中起作用。PCA图和相关性热图说明了组内重复样本的高度一致性及组间的染色质可及性的差异性。与来自未受伤心肌的心脏成纤维细胞相比,不同差异分组总计鉴定到100000个峰,其中36348个在MI后的心脏成纤维细胞中具有不同的可及性。
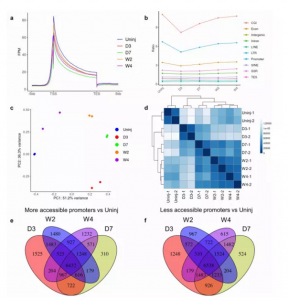 图3 不同分化状态心脏成纤维细胞的 ATAC-seq 分析
图3 不同分化状态心脏成纤维细胞的 ATAC-seq 分析
三、 差异基因表达与动态染色质可及性有关
为了研究启动子可及性是否与心脏成纤维细胞中的基因表达相关,将RNA-seq和 ATAC-seq数据结合起来进行分析(图4)。发现至少在一种状态下心脏成纤维细胞中表达的基因的启动子可及性高于任何状态下未表达的基因。相关性分析表明在30.77%的基因中发现启动子可及性和转录之间有着很强的正相关性,包括Tcf21(一种静止的心脏成纤维细胞标记物)、Ki67(一种增殖标记物)、Acta2(一种肌成纤维细胞标记物)和Comp(一种基质纤维细胞标记物)。
然后根据检测到的启动子的可及性对它们进行分类:高可及性组,低可及性组和动态可及性组。发现在三个组中,很多基因的启动子可及性和转录水平有很强的相关性,但是在高可及性组中有一些基因的启动子可及性和转录组水平有着较弱的或者无相关性。有趣的是,具有高启动子可及性的基因具有最高的CpG密度,而具有低启动子可及性的基因具有最低的CpG密度(图4g)。当根据启动子中的CpG密度将5,431个差异表达基因分为3组时,发现CpG密度较低但具有很强的正相关性的基因占比最高,进一步表明DNA甲基化可以独立调控基因转录,而与启动子染色质可及性无关(图4h)。
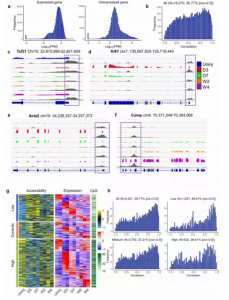 图4 差异基因表达和启动子可及性的相关分析
图4 差异基因表达和启动子可及性的相关分析
四、 启动子和远端区域的差异染色质可及性与差异基因表达相关
接下来选择了在启动子和远端区域具有差异可及性的基因,研究了它们在不同状态下心脏成纤维细胞中表达水平的变化。共计鉴定出3518个具有差异可及性的启动子,其中30.96%的启动子的可及性与基因表达呈高度正相关性(图5a,b)。这表明大部分具有差异可及性的启动子在其相关基因的表达中缺乏相应的变化。此外,与具有中等或高CpG密度的启动子相比,具有低CpG密度且表现出高度正相关的启动子占比最高,进一步表明启动子区具有较高的CpG密度的基因,更可能受DNA甲基化而不是启动子可及性的调节(图5b)。
总共确定了41,713个远端ATAC-seq峰,这些峰显示出不同的可及性。通过分析这些远端ATAC-seq 峰的可及性与其靶基因的表达之间的相关性,发现一些远端ATAC-seq峰有着高度正相关性,但是这些峰的占比低于启动子区域中可及性和基因表达呈正相关的峰的占比(图 5c,d)。此外,大多数远端 ATAC-seq 峰具有较低的CpG密度(图5d),表明DNA甲基化在TF和增强子之间的相互调节作用中可能没有起主要作用。
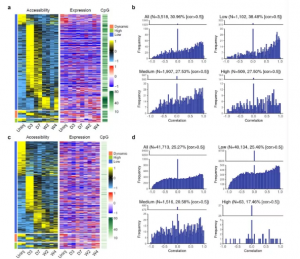 图5 启动子和远端区域的差异可及性和相应的基因表达变化
图5 启动子和远端区域的差异可及性和相应的基因表达变化
五、 MI后心脏成纤维细胞激活和分化的主要增强子和靶向TFs的预测
研究发现,TFs主要靶向远端区域的增强子,这在基因表达调控中起重要作用。在远端 ATAC-seq 峰中鉴定到440个已知motif,其中263个对应的TF 在心脏成纤维细胞中是有表达的。为了识别最有可能导致心脏成纤维细胞在MI后分化的motif和相应的TF,根据它们在不同状态的心脏成纤维细胞中的富集情况,将这些motif分为几个组(图 6a-e)。发现肌成纤维细胞中Rora motif的富集显著下降,并且在肌成纤维细胞和基质纤维细胞中都发现了Tcf21 motif富集程度在不断的逐渐减少(图 6b)。还发现一些motif(Nf1、Nfil3、Rela、Ets1 和 AP-1等TFs)的富集在肌成纤维细胞中暂时增加,然后在基质成纤维细胞中减少到接近或低于在静止心脏成纤维细胞中的富集水平(图 6c ,d)。
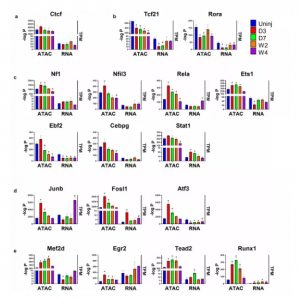 图6 远端ATAC-seq峰中TF motif的鉴定
图6 远端ATAC-seq峰中TF motif的鉴定
接下来分析每个分组中这17个TF motif在远端ATAC-seq 峰中的结合位点,发现与未受伤组相比,对应的靶基因在MI后的分组样本的心脏成纤维细胞中是上调的。分析结果说明除了Ctcf,大多数MI后的分组样本中的TF motif在远端ATAC-seq峰对应的上调靶基因区域中的富集程度高于所有远端ATAC-seq峰(图7a-e)。
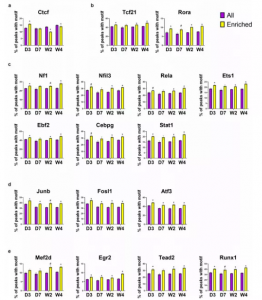 图7 远端ATAC-seq峰中TF motif的富集分析
图7 远端ATAC-seq峰中TF motif的富集分析
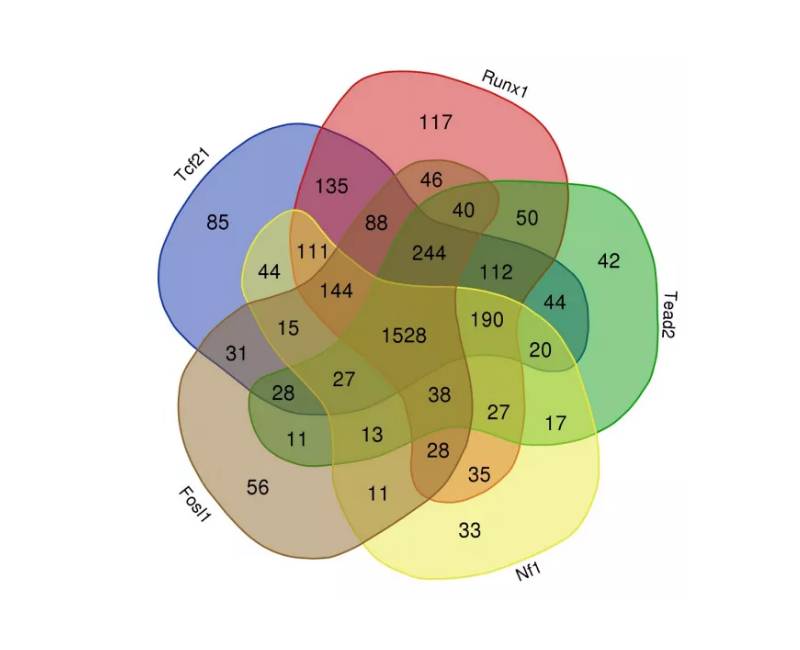
单个TF靶向的差异表达基因的比较结果揭示在Tcf21、Runx1、Tead2、Nf1 和 Fosl1 之间有着最大的交集(图 8),表明许多与MI后心脏成纤维细胞的激活和分化相关的基因是受多个关键TF共同调节的。
 图8 单个TF靶向基因的韦恩图
图8 单个TF靶向基因的韦恩图
六、 构建心脏成纤维细胞基因调控网络图
为了更好地了解这些motif及其相应TF在MI后心脏成纤维细胞基因表达变化中可能起了哪些调节作用,确定了七个选定的motif(Fosl1、Rora、Stat1、Nf1、Tead2、 Runx1 和 Tcf21)所结合的基因。在这些基因中挑选了在不同状态的心脏成纤维细胞之间差异最大的前500个基因,与编码7个选定TF的基因(共506个基因)构建基因调控网络图(GRN)(图9a)。506 个基因中有467个基因至少受1个TF调控,这表明这些TF影响了心脏成纤维细胞中的基因表达变化。有趣的是,Tcf21 和 Rora对大多数相关基因有着负调控作用。
对GRN中受7个TF调控的基因进行GO富集分析,发现一些与MI后心脏成纤维细胞生理和功能变化相关的GO生物学通路显著富集,并用于预测心肌梗死后心肌成纤维细胞的激活和分化中单个TF可能起的调节作用(图 9b)。研究发现 Fosl1、Stat1、Tead2 和 Runx1 可能通过促进促增殖基因表达或抑制抗增殖基因表达来刺激心脏成纤维细胞增殖。
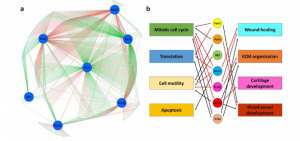 图9 心脏成纤维细胞中的基因调控网络图
图9 心脏成纤维细胞中的基因调控网络图
利用RNA-seq和ATAC-seq测序技术,发现在心脏成纤维细胞的不同状态下,启动子的可及性与许多基因的表达水平之间存在正相关,提示ATAC-seq是研究心肌梗死后成纤维细胞启动子可及性的有力工具。有趣的是,心脏成纤维细胞在不同状态下有相当数量的差异表达基因缺乏相应的启动子可及性变化,特别是在CpG密度高的基因中,这表明DNA甲基化可能在不影响启动子可及性的情况下调节启动子活性。暗示未来是需要对心肌梗死后心脏成纤维细胞激活和分化过程中DNA甲基化和染色质重塑的的协同和独立作用开展研究。本研究中motif富集分析和 GRN 分析的一个局限性是缺乏对 TF 与目标motif结合的验证以及验证这些 TF 功能的实验。本文研究揭示了一些关键TFs在心肌梗死后心脏成纤维细胞激活和分化中的可能作用,在未来的研究中,每一种都可以有针对性地研究,以更详细地探讨它们的功能作用。
总结
总的来说,通过转录组和开放染色质分析表明,染色质可及性可能在心肌梗死后心脏成纤维细胞的激活和分化中发挥重要作用。这一综合分析还表明,在心脏成纤维细胞的基因表达调控中,除了染色质可及性外,还有其他机制的存在。最后,motif分析和GRN构建工作确定了几个潜在的转录因子,它们可能通过对心脏成纤维细胞的转录组进行编程来协调介导心脏成纤维细胞的顺序激活和分化。
如果您对RNA-seq和ATAC-seq测序技术感兴趣,欢迎点击下方按钮联系我们,我们将免费为您设计文章思路研究方案。

参考文章:
Li C, Sun J, Liu Q, et al. The landscape of accessible chromatin in quiescent cardiac fibroblasts and cardiac fibroblasts activated after myocardial infarction [published online ahead of print, 2021 Oct 25]. Epigenetics. 2021;1-20. doi:10.1080/15592294.2021.1982158




 京公网安备 11011302003368号
京公网安备 11011302003368号