
百迈客宏基因组分析平台全新升级,百迈客宏基因组分析平台拥有流畅的主流程,丰富的个性化,在39种分析基础上,新增13种个性化分析,另还有6种个性化分析即将上线,满足您的定制化要求。
新增分析如下:
物种组成分析:新增upset图,厚壁菌门/拟杆菌门比例;
个性化功能注释:新增Ncyc数据库注释、分泌蛋白预测、T3SS效应蛋白预测、TCDB转运蛋白分类注释
Beta多样性分析:PLS-DA分析、分型分析
Alpha多样性分析:Alpha多样性指数、稀释曲线、香农指数曲线、等级丰度曲线相关性与关联分析:物种与功能贡献度分析
下面给各位老师介绍其中11项个性化分析内容,感兴趣的老师可以联系当地销售体验,另外转发本篇微信推文至朋友圈,集赞20+,保留12h以上,即可获得微生物多样性云分析一个月的免费体验 或 一次宏基因组(限6个样本)的标准分析和个性化分析体验权限。活动时间:2021.10.23-2021.11.2
宏基因组免费课程:http://live.biocloud.net/my/course/208
宏基因组分析平台总览
组成物种分析
1.upset图
维恩图(Venn diagram)是十九世纪英国数学家约翰·维恩(John Venn)发明,用于展示集合之间大致关系的一类图形。其中圈或椭圆重合(overlap)的部分就是集合与集合间元素的交集,非重叠部分则为特定集合的特有元素。集合图均可用于对集合共有和特有元素信息进行可视化,但是当数据分组过多(>4)时,维恩图看起来会非常杂乱,而集合图可以展示≥5个分组的集合元素共有和特有信息。统计样品之间的共有、特有元素。应用范围:适用于微生物、转录组、代谢组、单细胞等组学。如分析组间共有、特有物种;分析各比较组显著差异的共有、特有pathway/基因等。
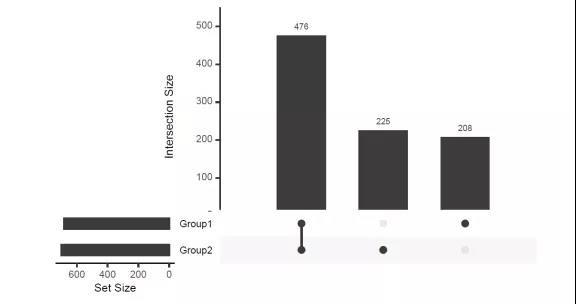
图1. upset示例,展示Group1/Group2两组组间共有和特有元素的数量
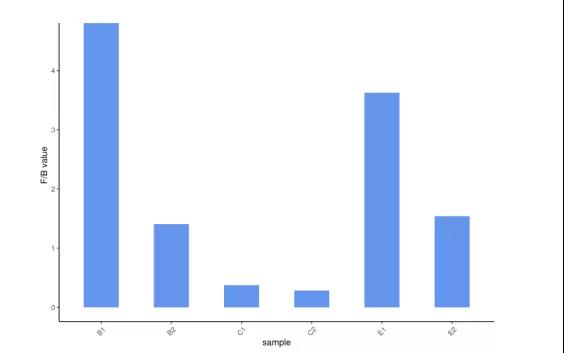
2.厚壁菌门/拟杆菌门比例
 图2 展示不同样本厚壁菌门/拟杆菌门比例
图2 展示不同样本厚壁菌门/拟杆菌门比例
个性化功能注释
1.Ncyc数据库注释
NCycDB(https://github.com/qichao1984/NCyc)是一个人工校正后的氮循环相关基因数据库。NCycDB 共包含 68 个基因(亚)家族,涵盖 8 个 N 循环过程,分别具有 95% 和 100% 同一性截止值的84,759和219,146个代表性序列,还确定了 1958 个同源直系同源组,并在数据库中包含了相应的序列,以避免由于“小数据库”问题导致的假阳性分配。NCycDB可用于快速而准确地对氮循环基因进行宏基因组学分析。
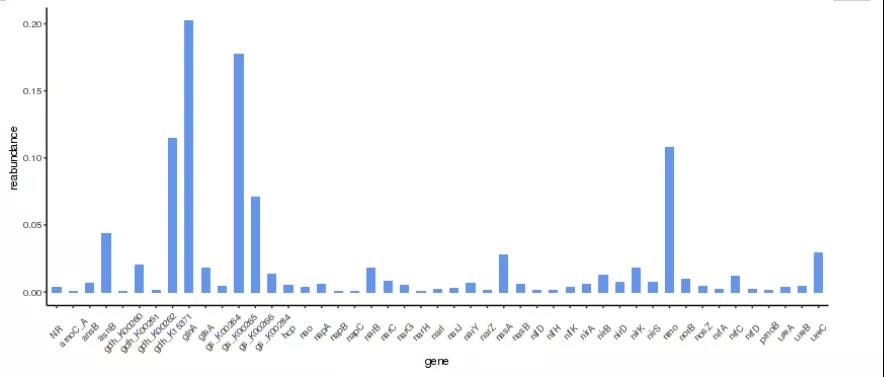
图3.Ncyc数据库注释
2. T3SS效应蛋白注释
三型分泌系统(type Ⅲ secretion system,TTSS,T3SS)是一个由多组分蛋白复合体形成的跨膜通道,它不依赖sec通过分泌蛋白或把这些毒力蛋白直接注入宿主细胞发挥致病作用,是许多革兰阴性致病菌中存在的复杂的分子装置。TTSS蛋白可以分成 4类:1)细胞膜结构装置蛋白 ( bacterialmembrane apparatus proteins),负责分泌系统组装;2) 转位蛋白 ( tranl ocon proteins ),在细胞膜上形成使效应蛋白得以通过的小孔;3)效应蛋白 ( translocated effect of proteins ),与病原的致病作用密切相关,能够引起宿主细胞相应的病理变化;4) TTSS分子伴侣 ( type Ⅲ chaper ones) ,能够有效地转移效应蛋白,保护其在未分泌之前不被降解。
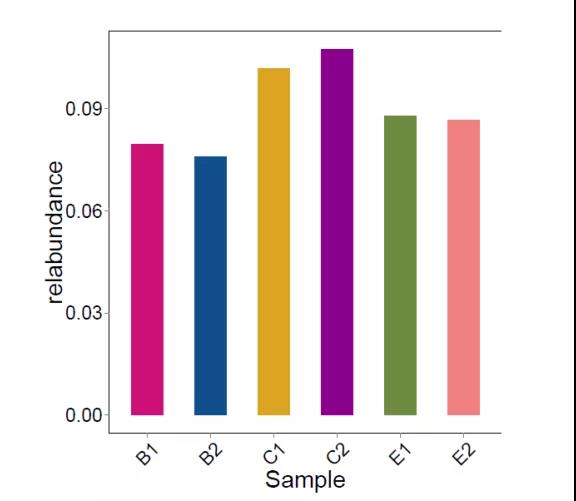 图4.T3SS效应蛋白注释
图4.T3SS效应蛋白注释
3. TCDB转运蛋白分类注释
TCDB(Transporter Classification Database)是包含各种转运蛋白的蛋白序列并对收录的蛋白进行分类的数据库。这个数据库的分类是依据于TC系统进行的分类(Transporter Classification system),具体的类别信息由TC号(TC number)标注,TC number由点分割的五组字符(D1.L1.D2.D3.D4)组成:D1(单个数字)代表相应的转运蛋白类(如通道蛋白、载体等);L1(单个字母)相应于转运蛋白亚类;D2(数字)代表相应的转运蛋白家族;D3(数字)代表相应的转运蛋白亚家族;D4(数字)相应于具体的转运蛋白本身。虽然在一些家族中有些转运蛋白可能利用多种作用模型或者可能利用不同于该家族中的其它蛋白的机制,但是对于大多数转运蛋白家族来说,家族中的成分具有相似的功能和作用机制。
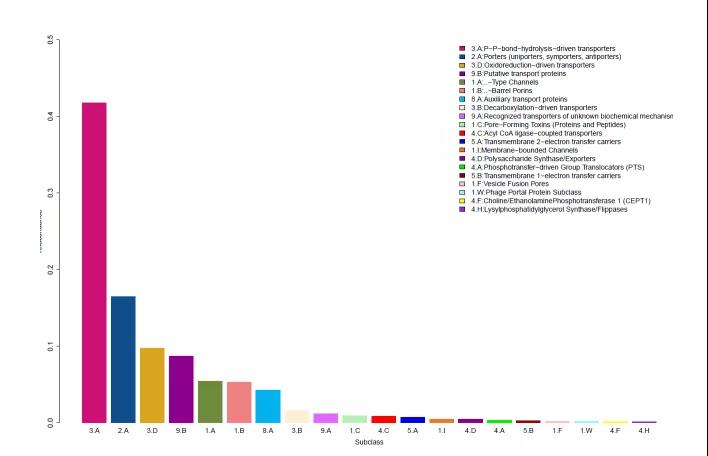 图5.TCDB转运蛋白分类注释
图5.TCDB转运蛋白分类注释
Beta多样性分析
1.PLS-DA分析
偏最小二乘判别分析(Partialleast squares discriminant analysis,PLS-DA)是多变量数据分析技术中的判别分析法,经常用来处理分类和判别问题。通过对主成分适当的旋转,PLS-DA可以有效的对组间观察值进行区分,并且能够找到导致组间区别的影响变量。PLS-DA采用了经典的偏最小二乘回归模型,其响应变量是一组反应统计单元间类别关系的分类信息,是一种有监督的判别分析方法。因无监督的分析方法(PCA)对所有样本不加以区分,即每个样本对模型有着同样的贡献,因此,当样本的组间差异较大,而组内差异较小时,无监督分析方法可以明显区分组间差异;而当样本的组间差异不明晰,而组内差异较大时,无监督分析方法难以发现和区分组间差异。另外,如果组间的差异较小,各组的样本量相差较大,样本量大的那组将会主导模型。有监督的分析(PLS-DA)能够很好的解决无监督分析中遇到的这些问题。
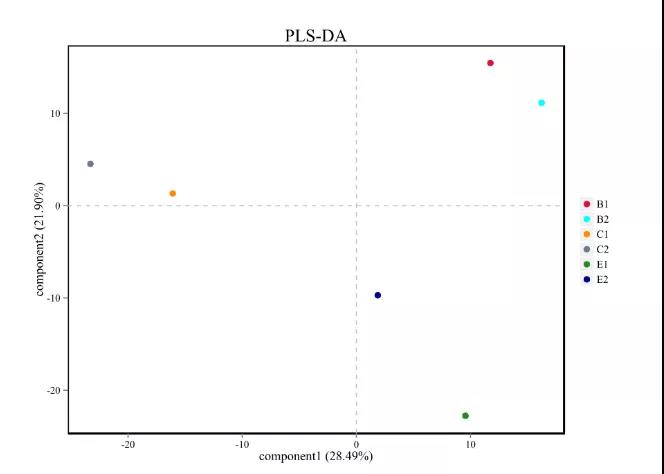
图6.PLS-DA分析
2.分型分析
分型分析,主要通过统计聚类的方法研究不同样本菌群物种/功能结构的分型情况。分型分析过程中一般不考虑环境因子等外部因素的影响。通过分型分析,可以将菌群物种/功能结构相似的不同样本聚为一类,主要适用于特定环境样本的菌群/功能分型,如肠道型(enterotypes)、口腔分型等。通常根据菌群在所选分类水平上的相对丰度,计算 Jensen-Shannon Distance和PAM (Partitioning Around Medoids)进行聚类,通过 Calinski-Harabasz (CH)指数计算最佳聚类 K 值,然后采用 Between-class analysis (BCA)或principal coordinates analysis (PCoA)进行可视化。
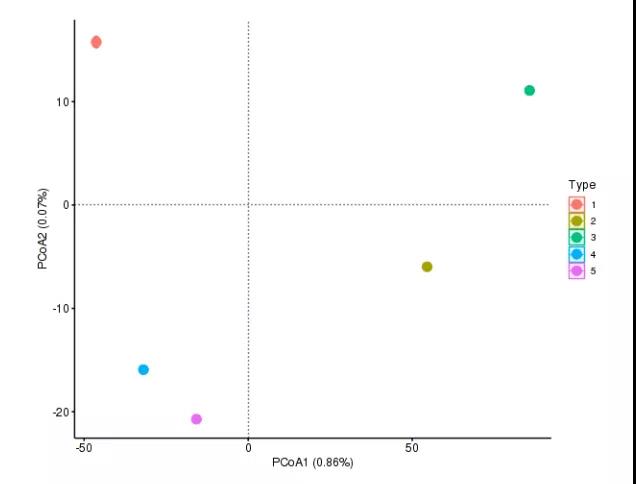
图7.分型分析
相关性分析
1.物种与功能贡献度分析
根据样本的物种、功能的对应关系,进行物种与功能相对丰度的关联分析,可以得到特定物种的功能贡献度、特定功能的物种贡献度。该分析既可以指定kegg、eggNOG、GO、Pfam、CAZy、CARD、VFDB、PHI-base、P450数据库的TOP/高丰度功能或代谢途径主要存在于哪些物种,也可以分析TOP/高丰度物种的主要功能或代谢途径。

图8.功能对物种贡献度图
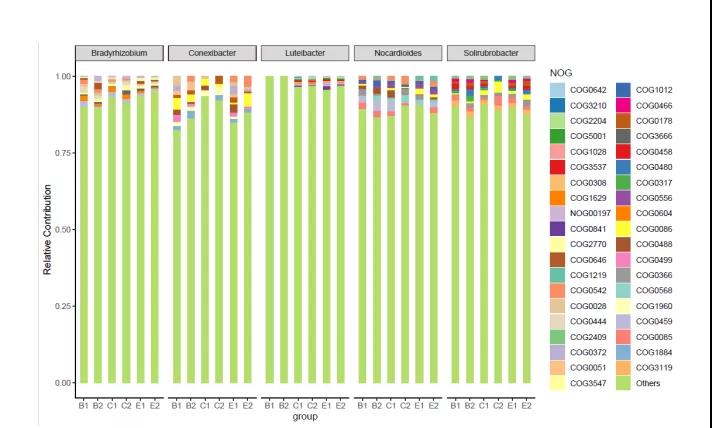
图9.物种对功能贡献度图
2、普式分析(即将上线)
Procrustes分析(Procrustes Analysis,普鲁克分析)是一种通过分析形状分布,比较两组数据一致性的方法。数学上来讲,就是不断迭代,寻找标准形状(canonical shape),并利用最小二乘法寻找每个对象形状到这个标准形状的仿射变化方式。该过程也称为最小二乘正交映射(least-squares orthogonal mapping)。Procrustes分析在面部几何中广泛应用,例如人脸识别。这里通过Procrustes Analysis分析物种组成和环境属性的潜在一致性。

图10.普式分析
3、VPA方差分解分析(即将上线)
VPA(Variance partitioning analysis)方差分解分析,可用于定量评估两组或多组(2~4组)环境因子变量对响应变量(如物种、功能组成)的单独解释度和共同解释度,常配合RDA/CCA使用。
分析软件:R语言vegan包中vpa分析。
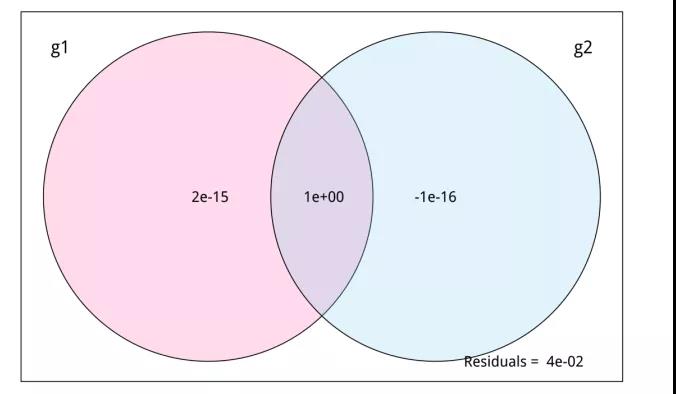
图11.VPA方差分解分析
4、环境因子排序回归分析(即将上线)
线性回归(Linear Regression)分析是利用数理统计中的回归分析,来确定两种或两种以上变量间相互依赖的定量关系的一种统计分析方法,在微生态研究领域运用十分广泛。基于样本对应的环境因子(pH、有机质含量、碳氮比等)和物种/功能丰度数据,分别对物种/功能的多样性进行计算,然后利用线性回归模型,对样本对应的环境因子数据与物种/功能的多样性数据进行线性回归分析,评估环境因子与物种/功能的多样性的一致性。当线性回归决定系数R2较高时,表明环境因子与物种/功能多样性的一致性较高。
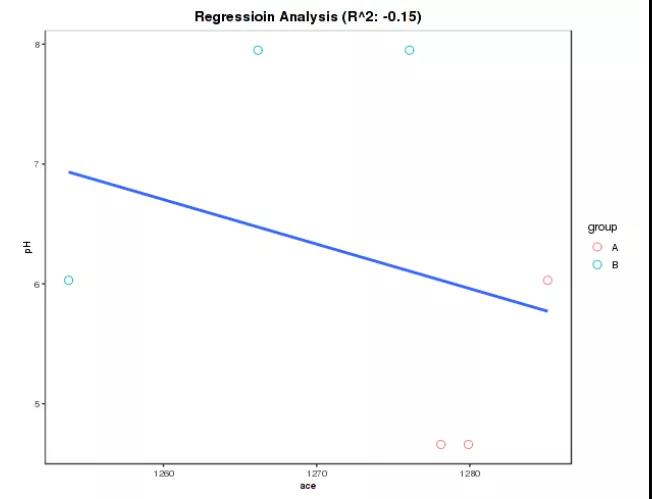
图12.环境因子排序回归分析
以上就是百迈客宏基因组分析平台已上线和即将上线的个性化分析内容,百迈客也将不断持续更新分析流程,满足您多样化的分析需求,为您的科研之路提供更便捷的测序及生信分析服务。




 京公网安备 11011302003368号
京公网安备 11011302003368号