
基因表达具有时间和空间特异性。scRNA-seq 使得我们可以在分辨率上研究转录组表达数据。空间转录组学(ST)可以将转录特征映射到不同组织区域,使得人们能够从空间的角度解析数据。百迈客可为您提供10x genomics scRNA-seq & spatial transcriptomics (ST) 技术服务,得到单细胞时空图谱数据实现在时空上分析基因的表达。
单细胞和空间转录组表达数据相关性分析
对同一样本的 scRNA-seq 的基因表达数据与空间转录组的基因表达数据或者同一样本的不同切片数据进行 Pearson 相关性分析,从而评估技术重复之间的相似性[1]。
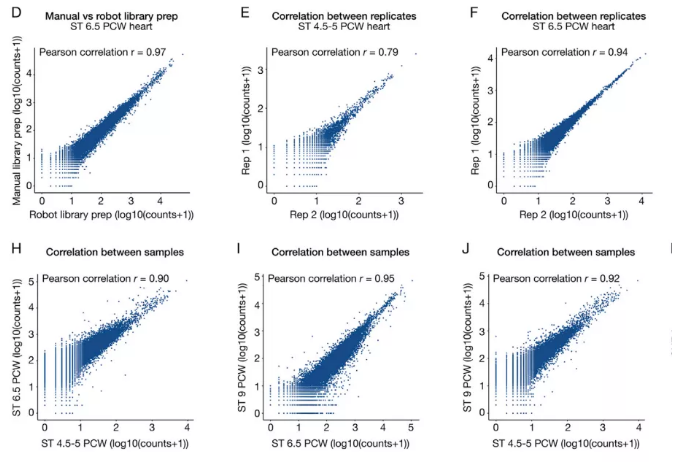 Fig1. 相关性散点图
Fig1. 相关性散点图
细胞类型和空间组织区域的锚定
将 scRNA-seq 数据集作为参考,使用因子分析来预测每个spot可能的单细胞组成,从而实现在空间上定位所有的 scRNA-seq 亚群[2]。
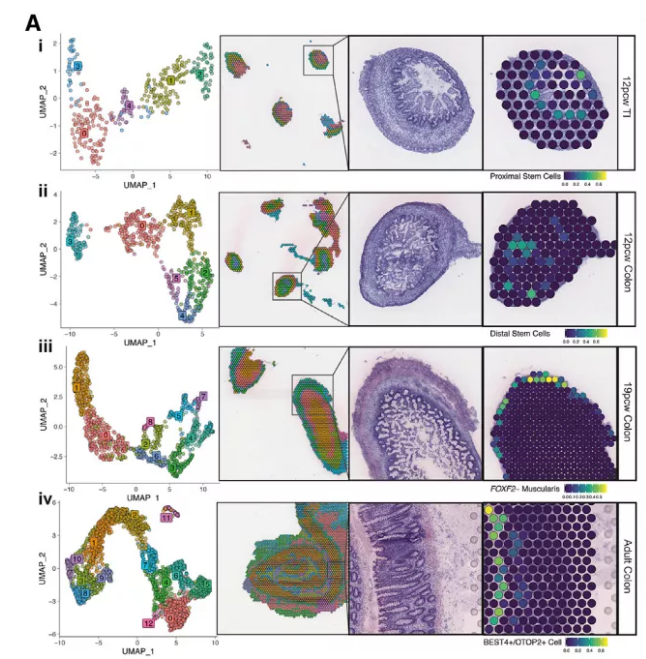
Fig2. 细胞类型在空间组织区域的分布
注:左侧图为ST的各个簇(cluster)的UMAP展示;左侧中间图为 clusters 在空间组织上的聚类可视化展示;右侧图为 ST 与 scRNA-Seq 细胞类型注释结果的整合
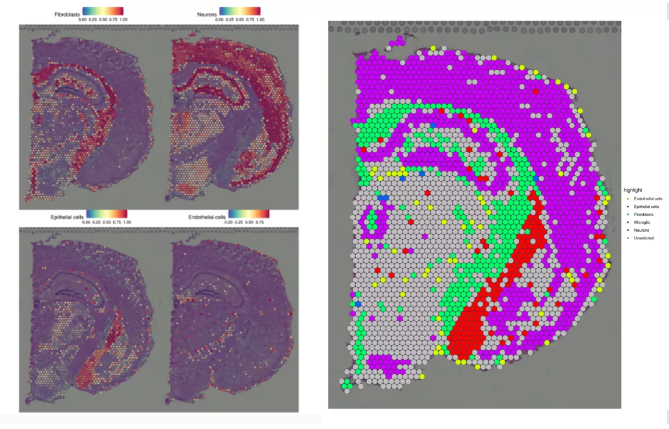
Fig3. 细胞类型在空间组织区域的分布
注:左图展示单独各个细胞类群在空间的分布情况,右图为空间组织切片每个spot的细胞类型展示图,不同颜色表示不同的细胞类型
多模式交互式分析
为了注释不同组织区域的√确的细胞组成,引入了 multimodal intersection analysis 方法[3]整合 scRNA-seq 数据和ST数据。MIA 方法通过计算某区域的差异基因与scRNA-seq数据鉴定的细胞类型差异基因之间的重叠程度,来推断特定组织区域中特定细胞类型的富集情况。
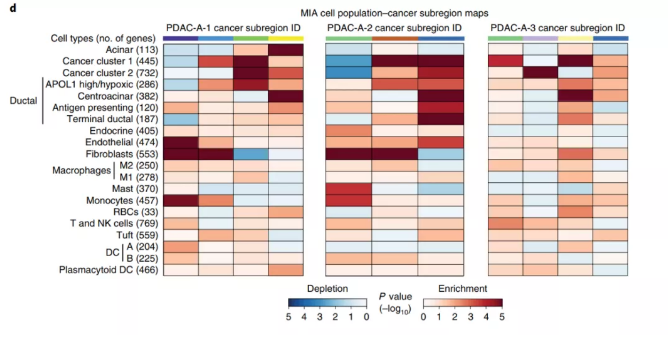
Fig4. MIA
注:热图行表示个细胞类型,列为空间组织区域,红色表示富集,蓝色表示缺失
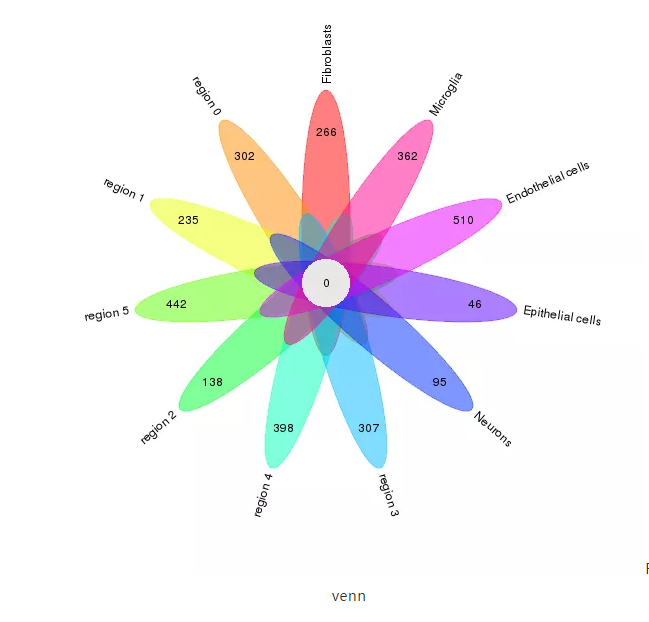
通过绘制韦恩图展示空间组织区域的差异基因与scRNA-seq各亚群差异基因的整体情况。

Fig5. venn
注:每个花瓣上的数字表示该细胞亚群或组织区域的差异基因总数,中间圆心的数字表示所有细胞亚群与组织区域的交集基因数
随着高通量测序技术的发展,scRNA-seq & ST 越来越多的被应用到各个领域的研究。但是如何实现scRNA-seq & ST 的联合,从更深的层面分析、挖掘数据?如何解析数据背后的生物学意义呢? 百迈客为您提供scRNA-seq & ST 服务分析平台,如果您也对单细胞和空间转录技术感兴趣,欢迎老师来电咨询!
参考文献
1. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart
2. Spatiotemporal analysis of human intestinal development at single-cell resolution
3. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma



 京公网安备 11011302003368号
京公网安备 11011302003368号