
此款工具基于DAVID的富集结果进行可视化分析,为客户解决了个性化展示GO/KEGG富集结果的需求,目前百迈客云平台上已经部署了相关的小工具,可助力客户实现相关需求。
在进行差异表达分析的时候,我们会获得大量的差异基因。就基于得到的差异基因,我们很难看出基因之间复杂的关系。为了更进一步了解这些基因的功能,富集分析在这一方面能够帮助我们。
GO是Gene Ontology的缩写,其中文我们翻译为基因本体论。到目前为止,GO的数据库由三大独立的Ontology建立起来:Biological Process生物过程、Molecular Function分子功能以及Cellular Component细胞组分,它们之间也具有联系。
Biological Process:细胞的每个部分和基因产物在细胞中的位置
Molecular Function:描述分子水平的活性
Cellular Component:由一个或多个分子功能有序组合而产生的生物学事件
GO的基本单位是term,每一个term对应一个属性。
KEGG是Kyoto Encyclopedia of Genes and Genomes的缩写,京都基因与基因组百科全书。
KEGG数据库是一个与通路相关的数据库,是大家最常见的代谢通路分析。
DAVID是一款免费的在线分析软件,我们只需要准备一列基因名(gene list)。下载其结果,如下图所示:
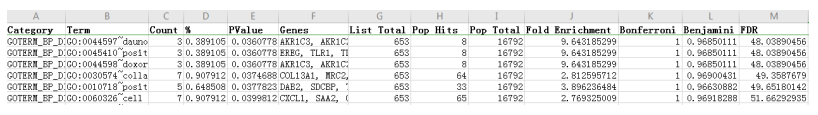
这与用R包跑出来的结果稍微有所不同,但都大同小异。
- (1)Count:即List-hits,也就是我们提交gene list中进入BP分类的某Term的数量
- (2)List total:提交gene list中进入BP分类的所有term的总数量
- (3)Pop hits:目前已被注释进入BP分类某Term的gene 数量
- (4)Pop total:目前已被注释进入BP分类的总gene数量
了解了这四个数值,计算出GeneRatio和富集因子,就可以利用ggplot对其进行可视化了,GeneRatio即注释在该条目中的感兴趣基因占所有差异基因数的比例;Rich.factor 富集因子,表示差异基因中注释到该通路的基因比例与所有基因中注释到该通路的基因比例的比值。富集因子越大,表示差异表达基因在该通路中的富集水平越显著。
data$GeneRatio <- data$Count / data$`List Total`
data$Rich.factor <- (data$Count / data$`List Total`)/(data$`Pop Hits`/data$`Pop Total`)
利用ggplot对其进行可视化
p = ggplot(data,aes(GeneRatio,Term))
p=p + geom_point()+theme(axis.text.x = element_text(colour=”black”,size=1))p=p + geom_point(aes(size=Count))pbubble = p+ geom_point(aes(color=Pvalue,size=Count))pr = pbubble+scale_color_gradient(low=”red”,high = “blue”)
pr = pr+labs(color=expression(Pvalue),size=”Count”,
x=”GeneRatio”,y=”Pathway”)
pr + theme_bw()
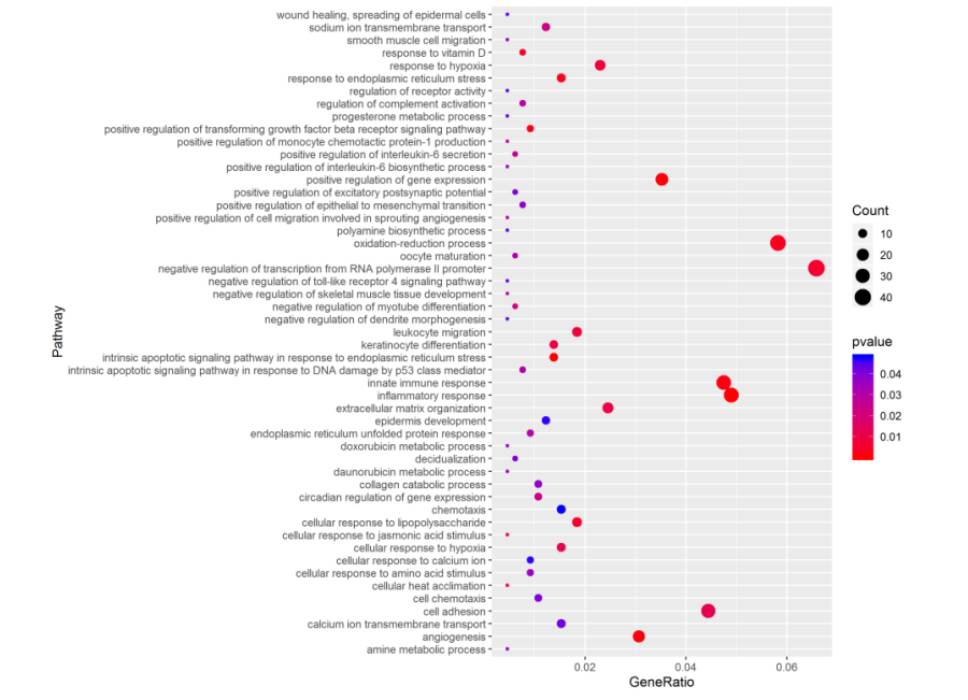
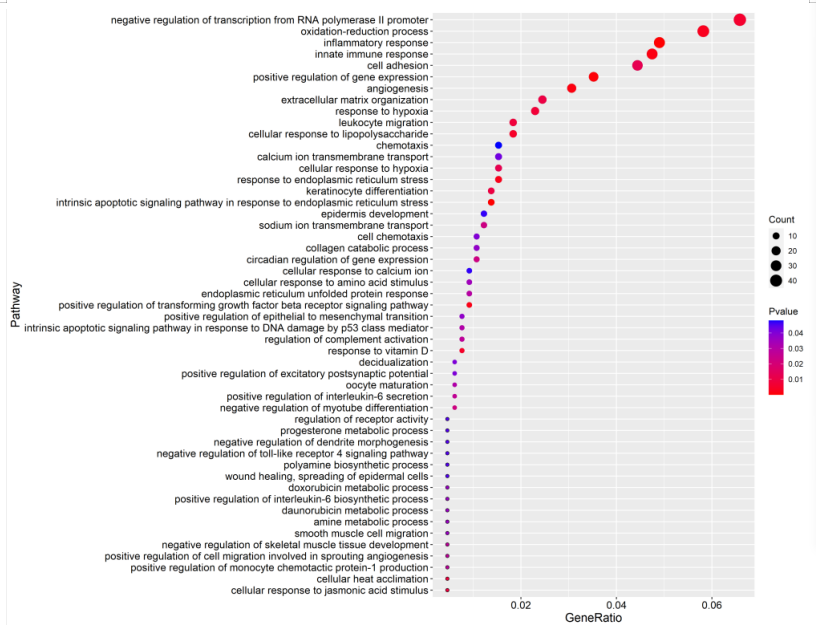
再对图片进行一下修饰,按照gene ratio大小排序画图,就可以得到一张整齐的气泡图啦,具体代码如下:
data <- data[order(data$GeneRatio),]
data$Term <- factor(data$Term,levels=data$Term)

点击按钮获取文章思路设计方案




 京公网安备 11011302003368号
京公网安备 11011302003368号