

基因组(Denovo sequencing),即基因组从头测序,指在不依赖参考基因组的情况下绘制该物种的全基因组序列图谱,从而获取该物种的全部遗传信息。高连续性基因组的获得,对后续功能基因定位,结构变异检测具有重要的意义。结合近几年的文章我们不难发现,基因组研究主要以下面几种方向为出发点开展:
1)大型/多倍体/超复杂物种基因组破译,技术创新改革;
2)0 Gap基因组/单体型基因组构建,序列优化打磨;
3)未知基因组破译联合多组学分析,经济价值挖掘;
4)品种泛基因组构建解析功能变异,覆盖多样表型;
5)科属水平谱系基因组构建与分析,探索进化功能;
6)多种基因组联合多组学比对剖析,解析性状特征。 … …
前5种好理解,第6种方向能做什么呢?其实我们想要了解一个物种,往往单一基因组难以完整解析,例如该物种不同性别如何引起性状差异?性别决定基因在哪里?例如表型明显差异的种质间的基因有哪些异同?例如两个物种表型相似但分类水平却存在争议?例如多倍体物种的演化历程模糊?等等棘手但是却又热门的研究话题。
接下来小编将通过百迈客最近三篇动植物上的成功案例带大家看看,如何通过数个材料基因组结合多组学的手段解析性状特征。
案例一

合作单位:中科院南海海洋研究所
发表期刊:Science Advances
影响因子:14.131
发表时间:2021.08
研究材料:
Denovo: 雌性与雄性草海龙(Phyllopteryx taeniolatus);雌性与雄性绿海龙(Syngnathoides biaculeatus)
个体重测序:2只雄性草海龙
RNA-seq:脑、眼、鳃、肝、肠、肌肉、鳍、皮肤和附叶
测序方案
Denovo:雌性、雄性草海龙与雄性绿海龙PacBio平台;雌性绿海龙Nanopore平台,雌性、雄性草海龙与雄性绿海龙进行Hi-C测序。三代测序技术对应测序数据如下表所示:
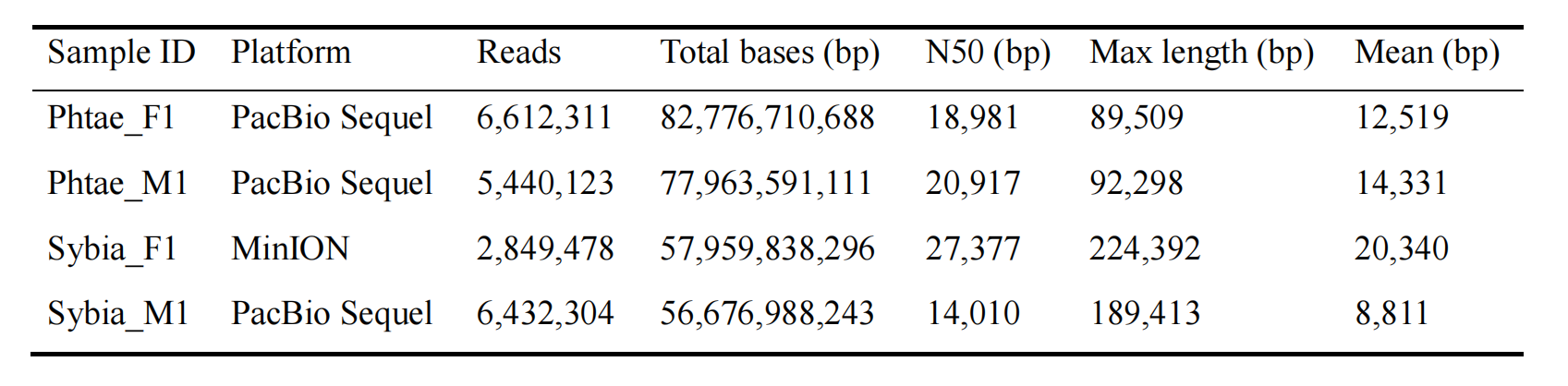
个体重测序:~30X PacBio
研究内容
1.两种海龙基因组组装与进化关系
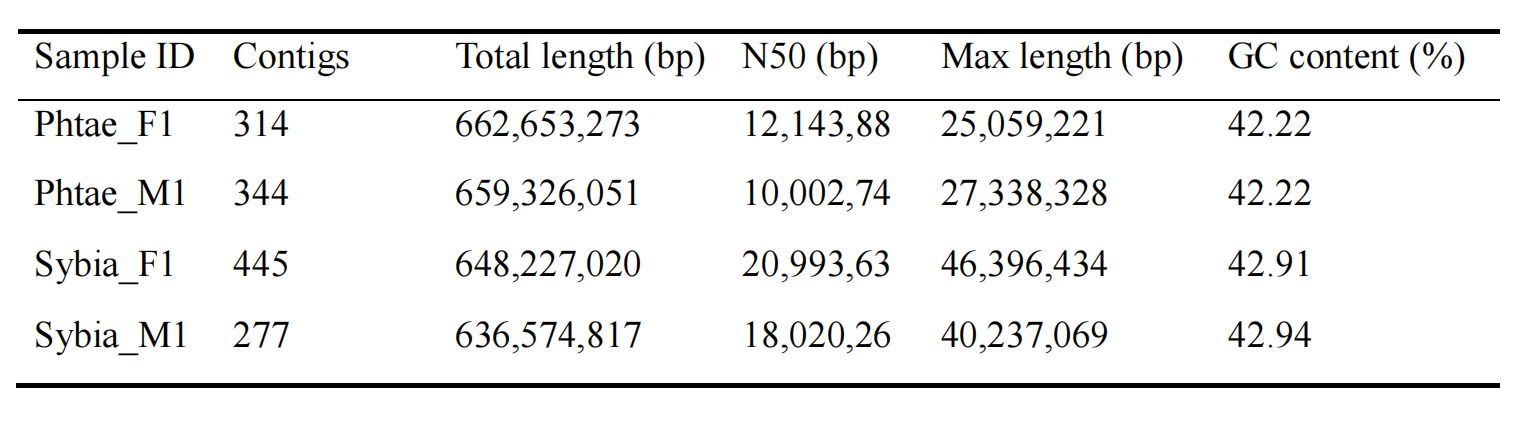
草海龙最终组装大小为 ~659 Mb(♂)与 ~663 Mb(♀), contig N50分别为10.0 Mb与12.1 Mb。绿海龙分别组装~637 Mb(♂)与~648 Mb(♀),contig N50分别为18.0Mb与21.0 Mb。4个基因组BUSCO评估显示范围在94.00- 94.40%。并分别在草海龙和绿海龙中确定了31个和33个发生扩张的基因家族。通过19条鳍鱼类全基因组数据集进行系统发育分析,明确草海龙与绿海龙在系统发育地位上属于海龙亚科(Syngnathinae)的姊妹群,并于27.3 百万年前左右发生分化。
2.“附叶”相关基因研究
草海龙的头部、颈部、腹部、背部和尾部区域有叶子状的附属物,可以与周围环境相融合,使草海龙以完美拟态隐匿于海草床中。这些结构是该物种的一种适应性进化产物,主要由骨基质和富含胶原纤维的结缔组织组成。通过转录组学分析,发现其表达基因(如msx, dlx, fgf)主要从皮肤和鳍等器官募集而来,暗示了相关基因对新器官产生和维持的重要作用。而“附叶”与鳍相比缺乏肢体发育特异性的hox基因。草海龙的附叶在捕食者的袭击中经常受到损伤,为了研究相关机制,作者通过转录组分析研究发现在其附叶中炎症和损伤修复相关基因表现出高表达水平,说明这些基因可能与其附叶的快速愈合和再生能力相关。同时草海龙特异性扩张的MHC I基因也在附叶中显著高表达,能为其提供额外的免疫保护。
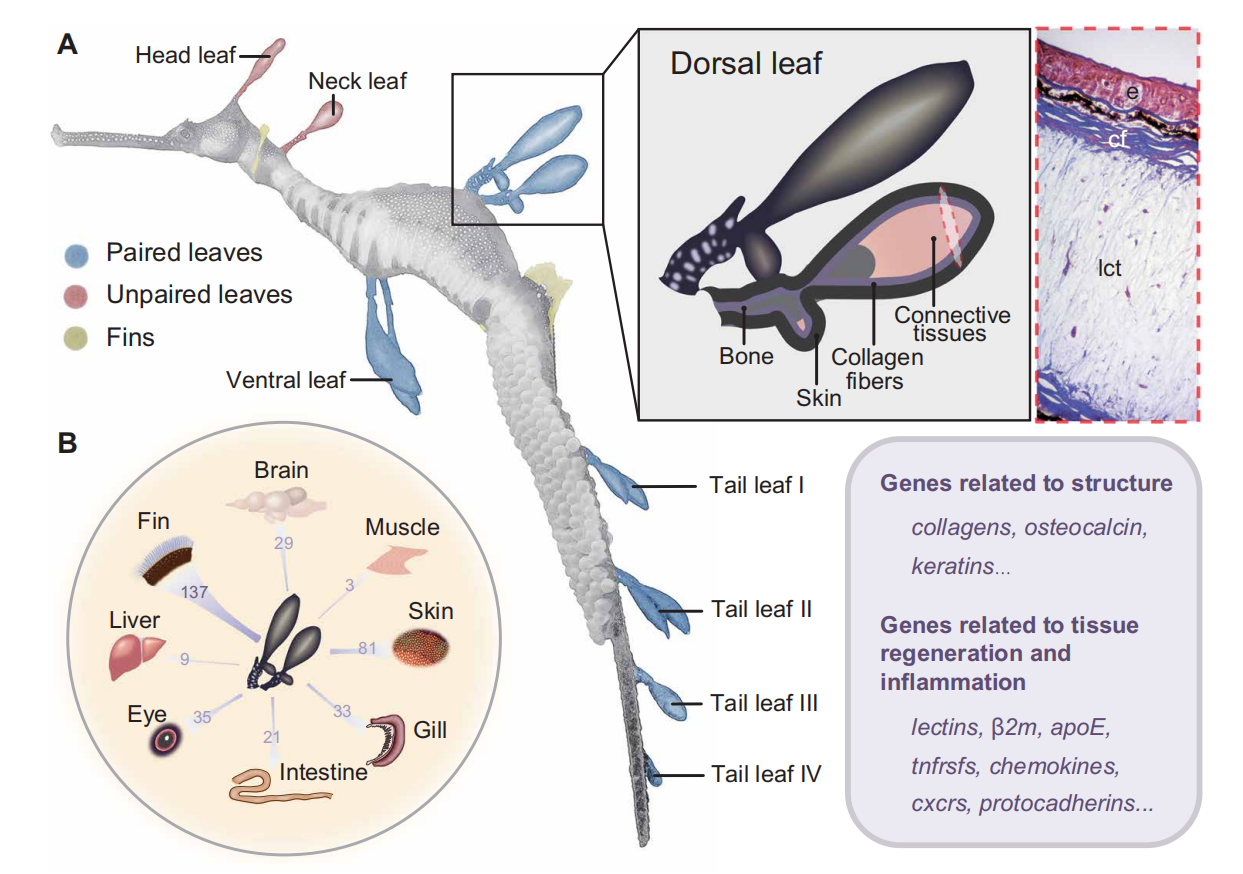
图1 草海龙附叶形态及关键基因表达特征
3.性别决定位点分析
通过雄性和雌性叶海龙Illumina reads正反比对雄性和雌性的全基因组序列,来确定叶海龙中假定的性染色体和性别基因座。结果显示Chr4上的一个~47-kb区域仅在雄性中存在, 且reads覆盖度为Chr4平均值的一半,该片段经Hi-C互作分析结果支持。注释及比较分析发现草海龙和绿海龙的性别决定基因均为amhr2的雄性特异性拷贝amhr2y,但两者的基因座不相同。系统发育分析表明,amhr2y起源于它们最近共同祖先的重复事件,而黄鲈amhr2y是从其谱系中的独立重复事件进化而来。研究发现amhr2y比amhr2受到的选择压力更强,其整体结构与amhr2相似。
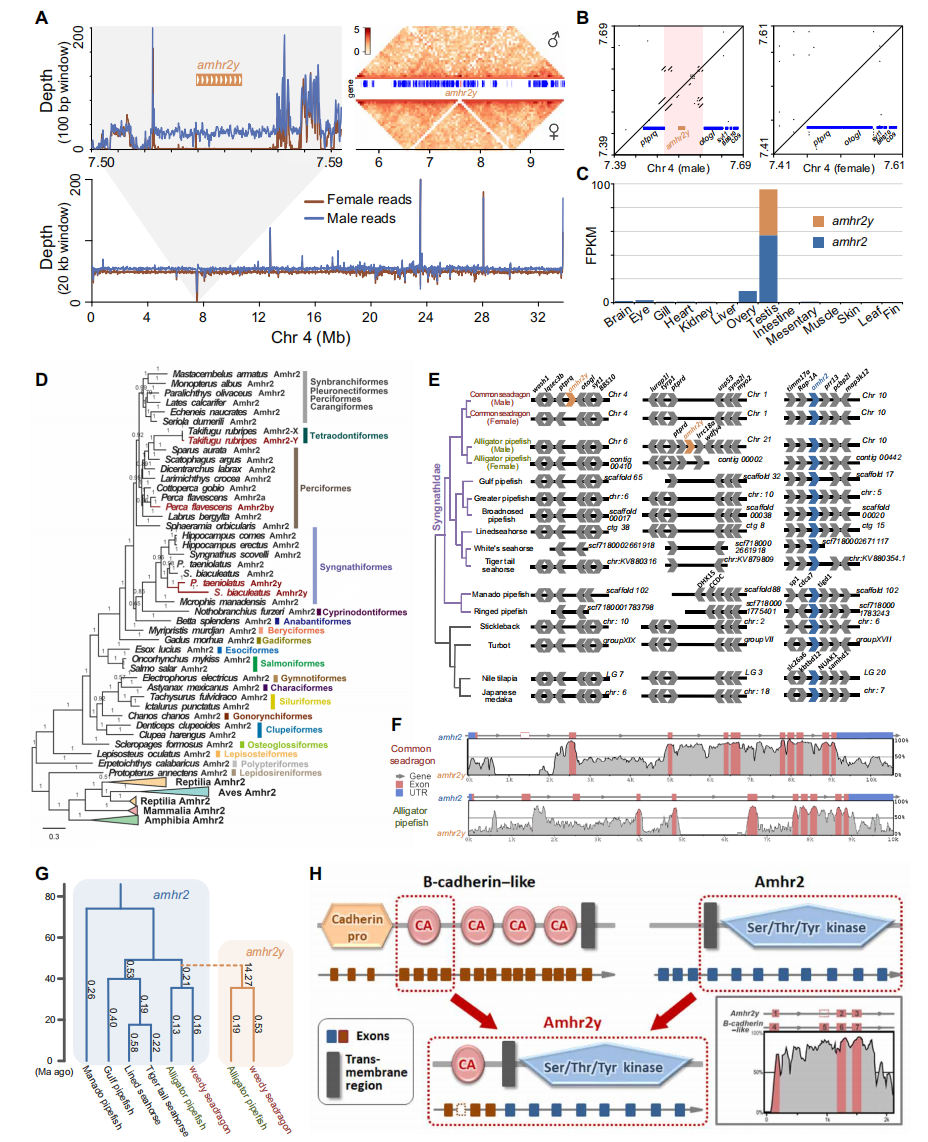
图2 草海龙与绿海龙性别决定基因进化
4.无牙研究
草海龙与其他海龙科物种一样具有缺乏牙齿的管状吻。研究表明,大部分富含 P/Q 的分泌型钙结合磷蛋白(SCPP)基因的缺失可能是导致syngnathids无牙的原因。为了验证海龙科中因假基因化丧失功能这一点,作者使用CRISPR-Cas9技术构建了两个斑马鱼scpp5突变系,发现scpp5-/-突变体斑马鱼牙齿的数量减少且颌骨中存在用于附着牙齿的凹坑。
研究结论
该研究通过雌雄性海龙基因组的破译,结合重测序分析、转录分析、比较基因组分析等研究揭示了海龙科物种性别决定基因的产生和演化历程,为海洋鱼类的环境适应性进化研究提供了重要理论依据。
案例二

合作单位:浙江大学
发表期刊:Plant Biotechnology Journal
影响因子:9.801
发表时间:2021.08
研究材料:
Denovo:Brassica juncea菜用芥菜T84-66、油用芥菜AU213;
个体重测序:12个油菜品种;
遗传进化:183份油用与菜用芥菜;
测序方案
Denovo:菜用芥菜分别146 Gb Illumina(~150X)+ 251 Gb PacBio(~200X)+Hi-C(~200X );油用芥菜147 Gb Illumina(~150X)+205 Gb PacBio(~200X)+Hi-C(~200X )
个体重测序:~20X Nanopore
遗传进化与GWAS:~10X illumina
研究内容
1.菜用芥菜T84-66与油用芥菜AU213基因组Denovo
本研究在首次完成榨菜基因组组装基础上(Nature Genetics,2016 【项目文章】NG芥菜基因组文章解读),进一步优化与解析低硫苷油用芥菜变种AU213的染色体水平基因组。调研图评估菜用与油用芥菜大小结果分别为968 Mb与938 Mb,且contigN50分别为3.36 Mb及4.4 Mb,*大的scaffold近乎跨越了完整的染色体序列。T84-66与AU213基因组BUSCO(97.7 %与98.3%)、CEGMA(99.6 %与99.8 %)、二代数据回比(97.12 %与96.18 %)、GOGGs验证以及与前期基因组版本比较等方式,共同表明本次基因组组装的高完整性。两种芥菜中分别预测了100,829及100,048个基因,与16年已发表的 T84-66 (V1) 版本相比有所增加。芥菜型油菜的Hi-C图谱显示常染色质(A)和异染色质(B)的分布对比显示,在着丝粒附近的异染色质状态中具有相对较低的基因表达模式。
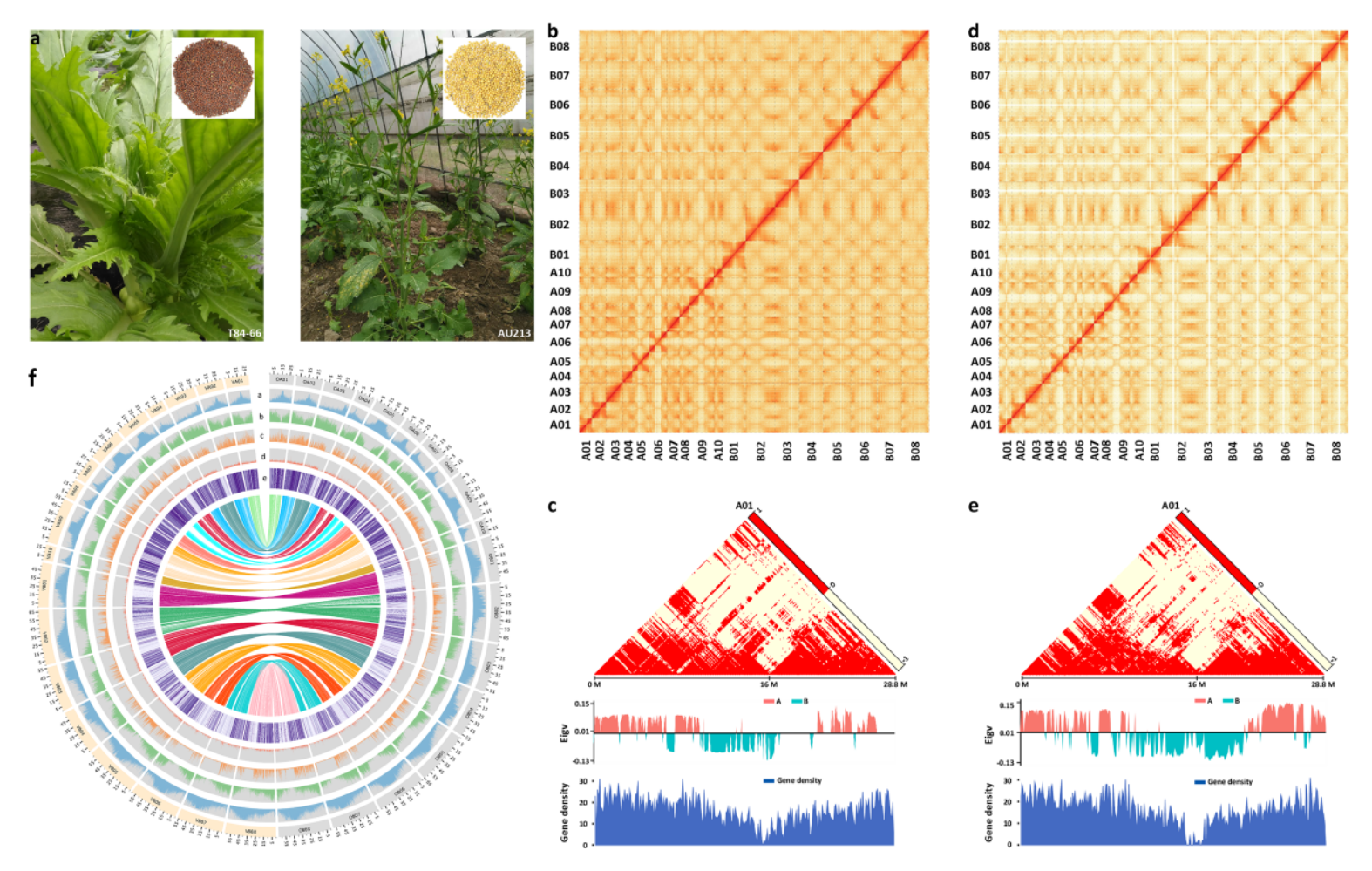
图1 芥菜表型与基因组特征以及三维基因特征
2.结构变异研究
系统地鉴定了T84-66 和 AU213 的A和B亚基因组中的全基因组单核苷酸多态性 (SNP)、插入/缺失 (InDels)和存在/缺失变异(PAV)。在T84-66和AU213之间的A和B亚基因组中鉴定了24,768个PAV(> 100 bp),其中3,634个PAV导致6,425个基因的变异。随机选择了几个PAV并使用PCR来确保这些PAV的保真度。其中一些基因组变异位于基因区域内,预计会影响T84-66和AU213作物中涉及生物和非生物胁迫的基因功能。
为了破译芥菜基因组菜用和油用品种之间SVs衍生的功能差异,作者基于Nanopore重测序技术,系统比较了菜用和油用芥菜群体基因组结构变异(structural variation,SV),挖掘到包括1, 354个高可信度的插入、缺失、重复、倒位、易位等变异。其中两个重要的基因位点TGA1和HSP20在ChrA06和ChrB08,可能与B.juncea基因组的菜用与油用品种之间对生物和生物应力的反应的自然变异有关。这些变异研究为菜用芥和油用芥两个典型分化群体的演化提供了基因组变异基础。
3.群体进化与GWAS分析
使用T84-66作为参考基因组,对183份油用与菜用芥菜进行进化关系分析,并通过SGS-GWAS(scored genomic SNPs based GWAS)基因定位,在A02和A09中发现了两个参与控制芥菜硫苷(GSL)积累变异的关键遗传位,并首次发现A09中的MYB28与B. jucnea中GSL的积累有关。经过进一步研究并同过ONT验证发现,MYB28基因的拷贝数变异(copy number variations,CNVs)是导致芥菜种群中硫苷积累差异的原因,该基因的拷贝数变异在低硫苷芥菜群体中普遍存在。
研究小结
该研究将为多倍基因组进化研究和√确基因组选择研究提供重要研究信息,对芥菜风味品质和油脂质量的分子遗传改良具有重要科学和应用价值。
案例三

合作单位:华中农业大学
发表期刊:Molecular Biology And Evolution
影响因子:16.241
发表时间:2021.05
研究材料:
基因组、Hi-C:圆叶棉G. rotundifolium(K2) 、亚洲棉G. arboreum(A2)、雷蒙德氏棉G. raimondii(D2)新鲜叶片
测序方案
denovo:illumina K2、A2和D5分别 108×, 118×, 132×;Nanopore K2、A2和D5分别124×, 131×, 167×
Hi-C挂载:6碱基酶HindⅢ;K2、A2和D5分辨率分别为20kb、20kb、10kb
Hi-C互作:4碱基酶DpnⅡ;分辨率20 Kb, 50 Kb, 100 Kb
研究内容
1.圆叶棉、亚洲棉、雷蒙德氏棉基因组组装注释
利用Nanopore测序技术组装了圆叶棉(K2)基因组,组装大小为2.44Gb(contigN50 = 5.33 Mb);提升了亚洲棉(A2)和雷蒙德氏棉(D5)的基因组,组装大小分别为1.62 Gb (contigN50 = 11.69 Mb)和0.75 Gb(contigN50 =17.04 Mb )。Hi-C挂载率均超过99%,BUSCO结果分别为 92.5%, 93.9%,及95.4%。
重复序列注释表明,相对于D5,K2和A2中棉种特异的反转录转座子扩增是造成这三个基因组大小三倍变化的原因, 特别是Gypsy和DIRS类型。全长转座子插入时间分析表明K2基因组中转座子插入*古老,A2基因组有更多新的转座子。
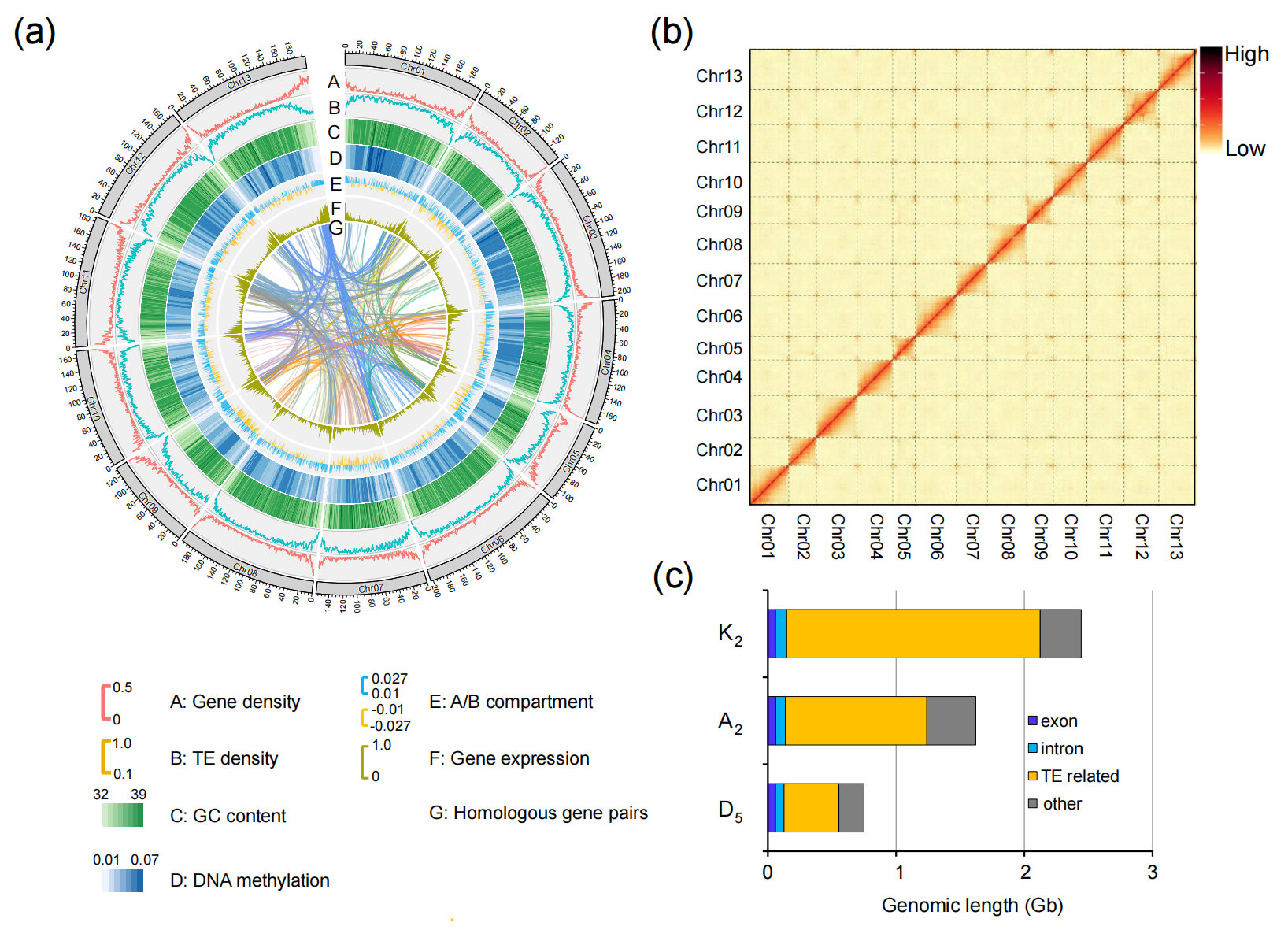
图1 圆叶棉基因组组装特征
2.比较基因组学和进化
比较基因组分析表明,A2和K2基因组在Chr01与Chr02染色体间存在一个大的易位;K2和D5基因组在Chr13与Chr05染色体间存在一个大的易位。三个棉种在57-71 百万年前存在一次共同的全基因组复制事件,并在5.1-5.4百万年前发生物种分化,基因共线性分析表明每个基因组大约有15%特异的基因家族。
3.A/B compartment演化
通过HiC染色质互作数据揭示三个棉种染色体大小的规律,A2与K2比D5多了约7000个基因,三个基因组中17%的共线性同源基因表现为A/B区室的染色质状态改变,这与活跃的转座子扩增相关。
K2与A2及与D5相比更多的倾向于A向B的转化。K2和A2中有更多的基因处于A compartment,D5中有更多的基因处于B compartment。
4.TAD结构演化及转座子扩增对TAD结构影响研究
大约60%的拓扑结构域(TAD)在三个基因组中发生了重新组织,K2基因组中有更多特异的TAD。基于边界TE覆盖度,边界TE表达以及TE插入时间分析,发现K2不保守的TAD边界存在特异的和较新的转座子(物种分化后爆发的TE)插入。这些结果表明最近在K2和A2基因组中表达的TEs的扩增可能有助于在三个物种分化后形成谱系特异性TAD边界。基于这些结果,作者提出了三个棉种分化过程中,基因组扩张-转座子扩增介导的A/B 区室转换和TAD重组的进化模型。
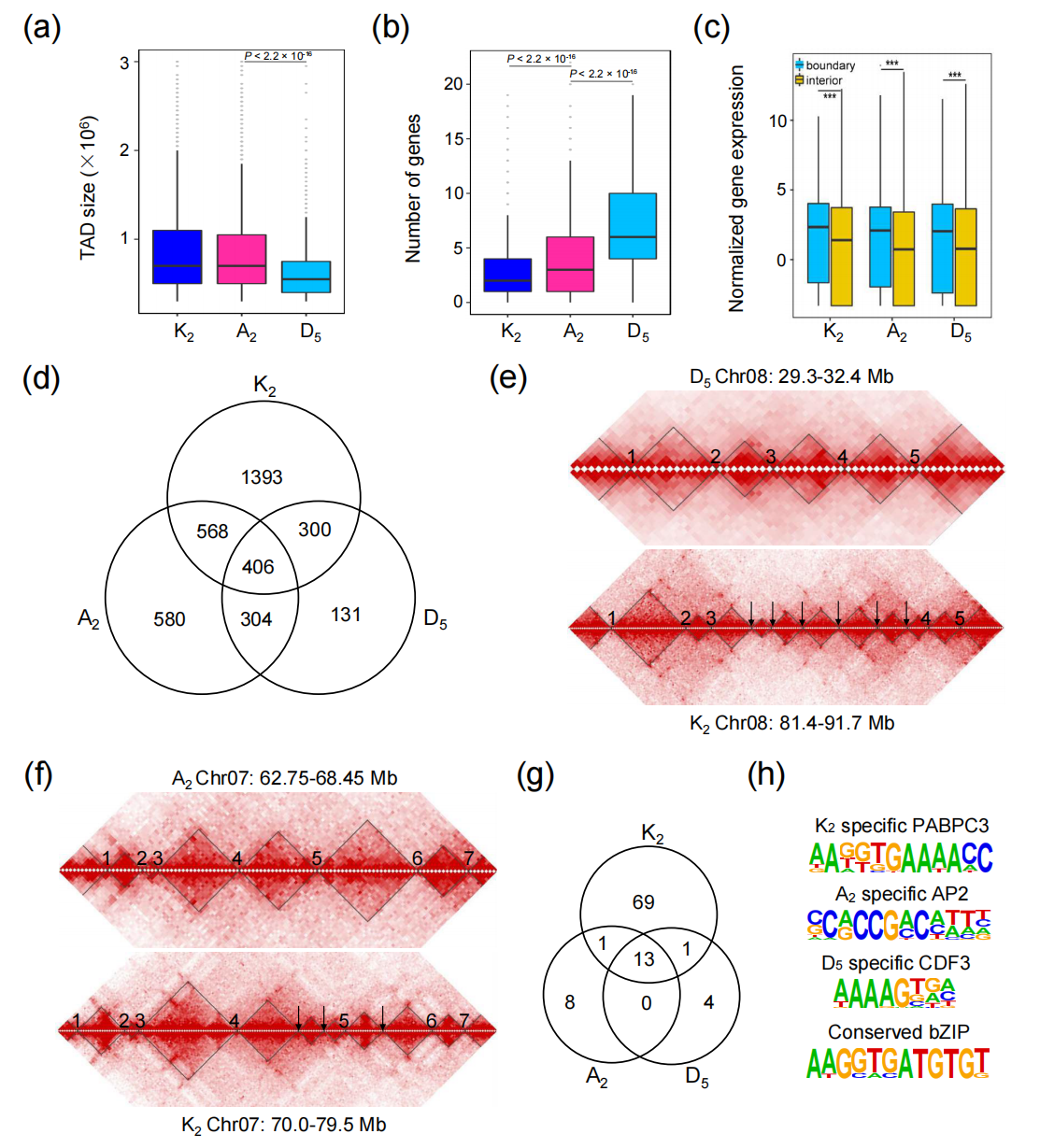
图2 三种棉TAD特征
研究小结
本次研究首次公布了棉属中二倍体圆叶棉基因组,并对亚洲棉和雷蒙德氏棉基因组进行了升级,解析了转座子活动驱动的基因组大小进化特征,从转座子扩增和染色质空间结构角度为棉花物种进化提供新的见解,为植物中转座子活动介导的转录调控进化研究提供参考。
百迈客2021年基因组成功案例展示



 京公网安备 11011302003368号
京公网安备 11011302003368号