
WGCNA简介
WGCNA(Weighted Gene Co-Expression Network Analysis, 加权基因共表达网络分析),是用来描述不同样品之间基因关联模式的系统生物学方法。可鉴定表达模式相似的基因集合(module),解析基因集合与样品表型之间的联系,绘制基因集合中基因之间的调控网络并鉴定关键调控基因。要了解 WGCNA, 首先需要理解什么是基因共表达网络。我们定义每个节点为一个基因,在不同样本中存在表达共性的基因处于同一个基因网络,而基因之间的共表达关系一般由它们之间的表达相关系数衡量。WGCNA 算法首先假定基因网络服从无尺度分布,并定义基因共表达相关矩阵、基因网络形成的邻接函数,然后计算不同节点的相异系数,并据此构建分层聚类树(hierarchical clustering tree),该聚类树的不同分支代表不同的基因模块(module),模块基因共表达程度高,而分属不同模块的基因共表达程度低。最后探索模块与特定表型或疾病的关联关系,最终达到鉴定基因网络的目的。
结果说明
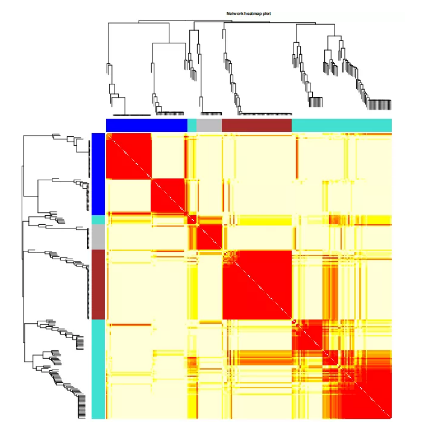
1. 基因共表达网络热图分析
WGCNA的核心步骤其实就是通过对TOM矩阵进行分层聚类,TOM (Topological overlap matrix)即为把邻接矩阵转换为拓扑重叠矩阵,以降低噪音和假相关,获得的新距离矩阵,可以用来计算基因之间关联程度。基于TOM矩阵,图A中的行和列均代表基因,灰色模块为无法分配到初级模块中的基因,树枝的顶端为核心基因,颜色越深代表TOM值越高。热图中方块的颜色越深(红)表示共表达相关性越高,越浅(黄)表示相关性越弱。
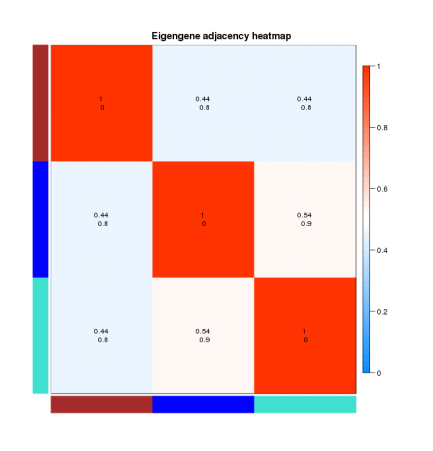
2. 模块间相关性热图
下图展示模块与模块间的相关性。横轴纵轴分别代表不同的模块,相关性越弱则呈现蓝色,相关性越强则呈现红色。
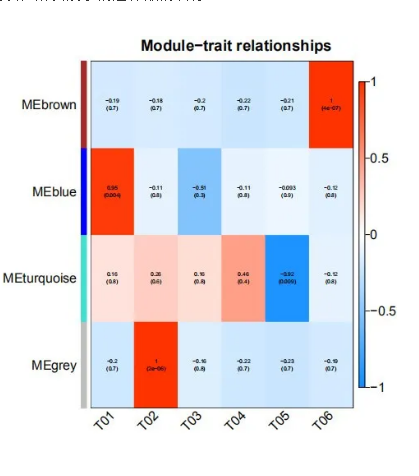
3. 模块与性状间的热图
模块与性状相关性热图:下图展示模块与给定性状的相关系热图。某一性状与某一模块的相关性越接近 1 的绝对值,很可能此性状与模块的基因功能相关。横轴代表着性状,纵轴代表着模块(下图中我们没有选择性状,所以展示的是样品的名称)。
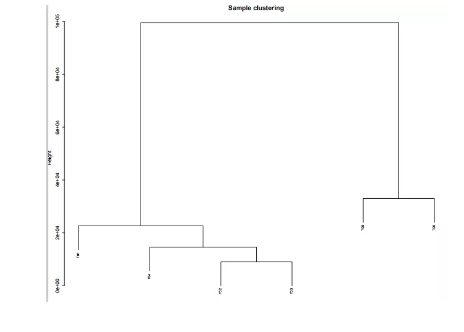
4.基因表达量的样本系统聚类树
基于基因表达量的样本系统聚类树: 在聚类中欧氏距离求两点的距离,聚类中用类平均法(average)计算集合之间的距离。同一枝上的样本具有更高的相似性。
5.样本系统聚类树及相关性状热图
在基因表达量的样本系统聚类树的基础上我们添加了热图,样本与性状之间的相关性越高热图中模块颜色也会越深。例如我们将热图左边的T01看作是性状标签,它与分支样本TP01的热图模块呈现红色,说明性状T01与样本T01的相关性非常高。
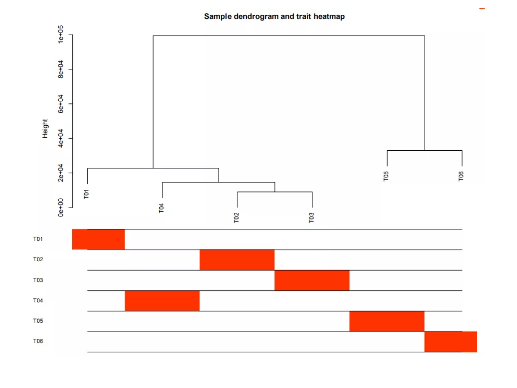
6.基因的系统树图及性状相关性热图
基因的系统树图及性状相关性热图主要分为三个部分:第一个部分为基因聚类绘制的聚类树,分枝代表基因;第二部分为基因聚类树根据不同的聚类对应到的不同模块;第三部分为性状的热图,如图中T01~T06为性状,对应热图中的颜色越深,说明该模块中的基因与该性状的相关性越高。
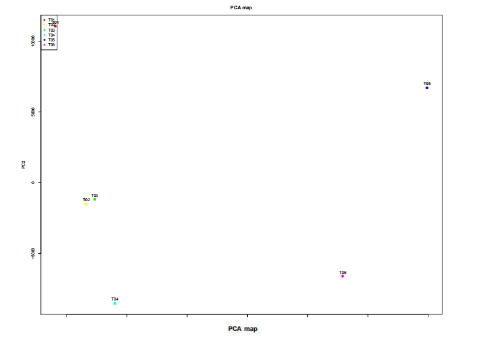
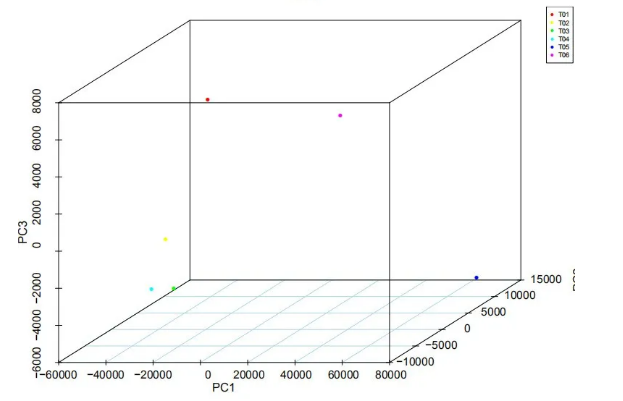
7.样品 PCA 分析二/三维图
PCA分析横坐标代表着第一主成分,纵坐标代表第二主成分,可以根据图看出在第一主成分方向上样本的距离越近,说明它们的主成分越相近,相关性越高。
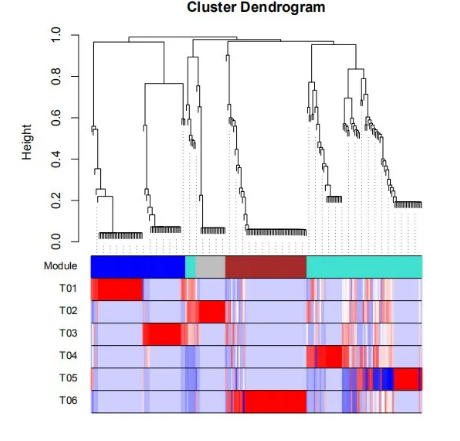
8.基因网络模块
基因网络模块主要分为三个部分:第一部分为基因的聚类树;第二部分为根据基因聚类树划分的不同模块;第三部分为根据第二部分模块进行了矫正,最后结果为校正后的模块。
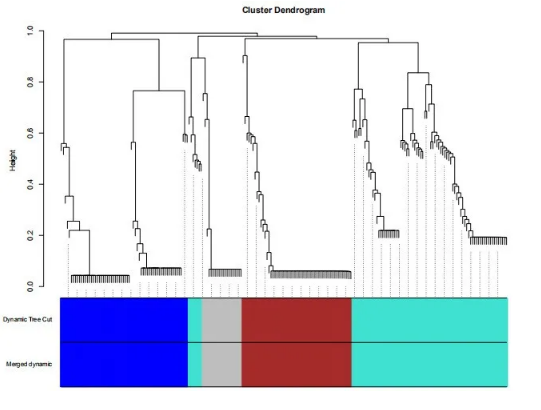
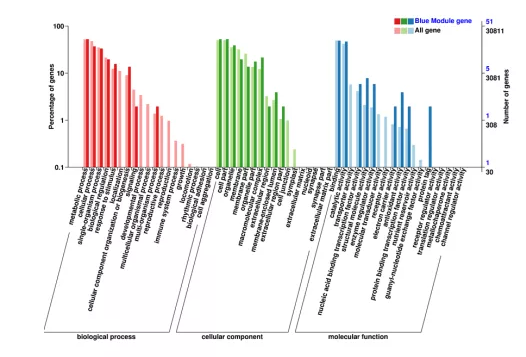
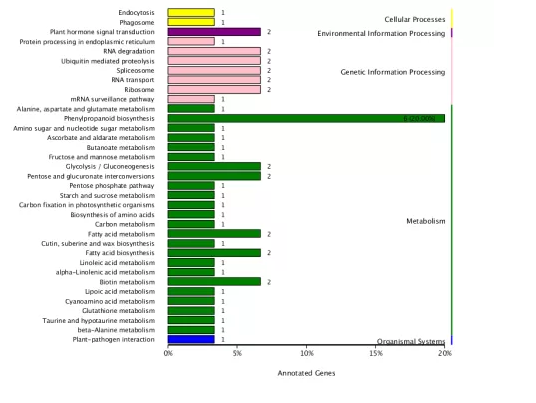
9.模块COG/KEGG/GO二级分类图
将每一个模块中的基因绘制了跟它功能相关的COG/KEGG/GO分类富集图。
在COG富集分析中横轴代表功能分类,纵轴代表基因频数。
在GO富集分析中横轴代表GO term,左侧纵轴代表基因占比,右侧为基因的数目。
在KEGG富集中纵轴代表路径,横轴代表注释到的基因数目

GSEA介绍
基因集富集分析(GSEA: Gene Set Enrichment Analysis),可以在没有先验经验存在的情况下根据所有基因表达情况对所有基因进行富集分析。一般的差异分析通常只集中关注于一些显著的上调或下调基因,而这会遗漏部分差异表达不显著却有重要生物学意义的基因。而GSEA不会设置差异阈值,能够检测出微弱但是一致的趋势。其输入数据包含两部分:一为已知功能的基因集 (可以是GO注释、MsigDB的注释或其它符合格式的基因集定义);二是表达矩阵 (也可以是排序好的列表)。
软件会对基因根据其与表型的关联度从大到小排序,然后判断基因集内每条注释下的基因是否富集于表型相关度排序后基因表的上部或下部,从而判断此基因集内基因的协同变化对表型变化的影响。
结果说明
百迈客中的GSEA分析采用KEGG通路以及GO的BP、CC、MF分支的基因集作为感兴趣的基因集合,以每个差异分组的log2FC作为背景基因集的打分来分析感兴趣基因集合的富集情况,最后控制pvalue<0.001,FDR 图片
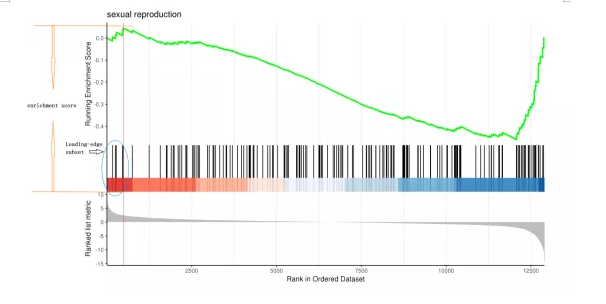
GSEA计算中几个关键概念
1、计算富集得分 (ES, enrichment score)。ES反应基因集成员(差异基因)在排序总基因列表的两端富集的程度。计算方式是,从总基因集的第一个基因开始,计算一个累计统计值。当遇到一个落在差异基因里面,则增加统计值。遇到一个不在差异基因里面,则降低统计值。
2、每一步统计值增加或减少的幅度与基因的表达变化程度(更严格的是与基因和表型的关联度,可能是fold-change,也可能是pearson corelation值,后面有介绍几种不同的计算方式)是相关的,可以是线性相关,也可以是指数相关 (具体见后面参数选择)。富集得分ES最后定义为峰值。正值ES表示基因集在列表的顶部富集,负值ES表示基因集在列表的底部富集。
3、评估富集得分(ES)的显著性。通过基于表型而不改变基因之间关系的排列检验 (permutation test)计算观察到的富集得分(ES)出现的可能性。若样品量少,也可基于基因集做排列检验 (permutation test),计算p-value。
4、多重假设检验校正。首先对每个差异基因子集计算得到的ES根据基因集的大小进行标准化得到Normalized Enrichment Score (NES),随后针对NES计算假阳性率。(计算NES也有另外一种方法,是计算出的ES除以排列检验得到的所有ES的平均值)Leading-edge subset为对富集得分贡献大的基因成员。
从前文中我们了解到GSEA分析的目的是要判断差异集基因(基于先验知识的基因注释信息,某个关注的基因集合)中的基因是随机分布还是聚集在排序好的总基因集的顶部或底部(这便是富集分析)。与GO富集分析的差异在于GSEA分析不需要指定阈值(p值或FDR)来筛选差异基因,我们可以在没有经验存在的情况下分析我们感兴趣的基因集,而这个基因集不一定是显著差异表达的基因。GSEA分析可以将那些GO/KEGG富集分信息中容易遗漏掉的差异表达不显著却有着重要生物学意义的基因包含在内。

蛋白网络互作
蛋白质互作网络(protein protein interaction network,PPI network)是由蛋白通过彼此之间的相互作用构成,来参与生物信号传递、基因表达调节、能量和物质代谢及细胞周期调控等生命过程的各个环节。系统分析大量蛋白在生物系统中的相互作用关系,对了解生物系统中蛋白质的工作原理,了解疾病等特殊生理状态下生物信号和能量物质代谢的反应机制,以及了解蛋白之间的功能联系都有重要意义。在生物医药领域有助于从系统的角度研究疾病分子机制、发现新药靶点等等。
主要主要是基STRING数据库提取目标基因集的互作关系。对于数据库中未收录信息的物种,使用BLAST软件,将目的基因与数据库中的蛋白质进行序列比对,寻找同源蛋白,根据同源蛋白的互作关系对构建互作网络。构建完成的蛋白质互作网络可导入Cytoscape软件进行可视化。
图片
结果说明
如图中的节点为蛋白质,边为互作关系。互作网络中节点(node)的大小与此节点的度(degree)成正比,即与此节点相连的边越多,它的度越大,节点也就越大。节点的颜色与此节点的聚集系数(clustering coefficient)相关,颜色梯度由绿到红对应聚集系数的值由低到高,聚集系数值越高表示此节点的邻接点之间的连通性越好。边(edge)的宽度表示此边连接的两个节点间的互相作用的关系强弱,互相作用的关系越强,边越宽。没有的组合代表没有互作关系。
图片
百迈客转录组测序服务针对不同样品类型采用全面解析mRNA信息的策略,不仅能获得样本的转录本结构和转录丰度信息,还能探究相关基因结构变异(SNP、CNV、SV、 Indel等)。此外,百迈客云的生物大数据分析平台,可以深入挖掘差异表达基因,完成多种高级分析内容,例如上文提到的WGCNA、GSEA、PPI等内容,为您提供全面、高效的生物信息分析以及整合利用公共数据库的解决方案。
欢迎点击下方按钮联系我们,我们将免费为您设计文章思路方案。





 京公网安备 11011302003368号
京公网安备 11011302003368号