
安装monocle 包
monocle 分析之数据导入
monocle 分析之细胞聚类
数据预处理降维、可视化
去批次效应后重新降维
monocle3可以分析大型、复杂的单细胞数据,可处理数百万个细胞。monocle3主要更新了哪些功能?
1.一个更好的结构化工作流程来学习发展轨迹。
2.支持 UMAP 算法初始化轨迹推断。
3.支持具有多个root的轨迹。
4.学习具有循环或收敛点的轨迹的方法(Ways to learn trajectories that have loops or points of convergence)。
5.利用“近似图抽象”的思想,⾃动划分细胞以学习不相交或平⾏轨迹的算法。
6.一种新的对轨迹依赖表达的基因的统计测试。这将替换旧的differalgenetest()函数和BEAM()。
7.3D界面可视化轨迹和基因表达。
安装monocle3包
要求R > 3.6.1
1、安装Bioconductor
1 if (!requireNamespace(“BiocManager”, quietly = TRUE))
2 install.packages(“BiocManager”)
3 BiocManager::install(version = “3.10”)
2、安装依赖包
1 BiocManager::install(c(‘BiocGenerics’, ‘DelayedArray’, ‘DelayedMatrixStats’,
2 ‘limma’, ‘S4Vectors’, ‘SingleCellExperiment’,
3 ‘SummarizedExperiment’, ‘batchelor’, ‘Matrix.utils’))
3、安装monocle3
1 install.packages(“devtools”)
2 devtools::install_github(‘cole-trapnell-lab/leidenbase’)
3 devtools::install_github(‘cole-trapnell-lab/monocle3’)
如果不能联网下载,可以下载到本地然后本地安装
1 library(devtools)
2 install_local(“D:/monocle3-0.2.2.tar.gz”)
monocle3分析之数据导入
1、从seurat对象生成 cell_data_set (CDS)对象
.expression_matrix , a numeric matrix of expression values, where rows are genes, and columns are cells
.cell_metadata , a data frame, where rows are cells, and columns are cell attributes (such as cell type, culture condition, day captured, etc.)
.gene_metadata , an data frame, where rows are features (e.g. genes), and columns are gene attributes, such as biotype, gc content, etc.
注:表达矩阵的⾏数必须与gene_metadata相同,表达矩阵的列必须与cell_metadata相同
1 library(monocle3)
2 sceObj <- readRDS(“/path/sce_test.Rds”)
3 expression <- GetAssayData(sceObj, assay = ‘RNA’, slot = ‘counts’)
4 cell_metadata <- sceObj@meta.data
5 gene_annotation <- data.frame(gene_short_name = rownames(expression))
6 rownames(gene_annotation) <- rownames(expression)
7 monocle_cds <- new_cell_data_set(expression, cell_metadata = cell_metadata, gene_metadata = gene_annotation)
2、直接从10X cellranger生成CDS对象
monocle_cds 注:也可以使⽤ load_mm_data() 函数读取 MatrixMarket 格式数据
monocle3分析之细胞聚类
我们使用monocle3对数据进行聚类分析。
数据预处理
使用 preprocess_cds 对数据进行normalize 以及 PCA 降维
monocle_cds <- preprocess_cds(monocle_cds, num_dim = 50)
注:可以使⽤ plot_pc_variance_explained() 函数画肘部图,从中选择合适的 PCs 数降维、可视化
1 monocle_cds <- reduce_dimension(monocle_cds, preprocess_method = “PCA”) # 默认使⽤UMAP
2 plot_cells(monocle_cds, color_cells_by=”seurat_clusters”) # umap显示原seurat 细胞聚类的结果
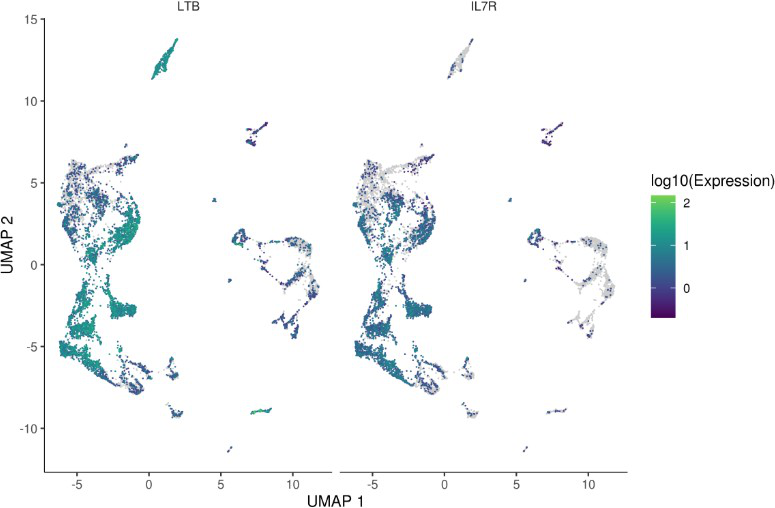
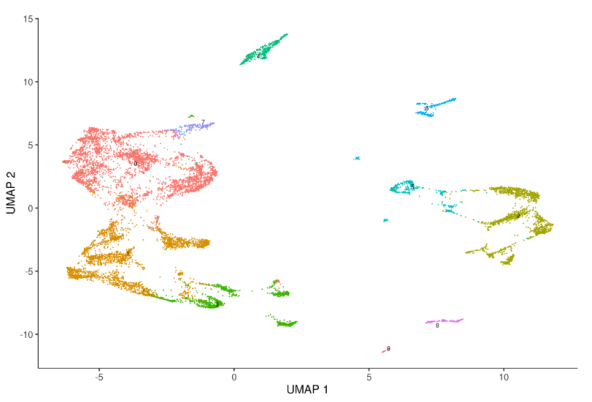 plot_cells(monocle_cds, genes=c(“LTB”, “IL7R”)) # 显示某几个基因的表达
plot_cells(monocle_cds, genes=c(“LTB”, “IL7R”)) # 显示某几个基因的表达
去批次效应后重新降维
检查样本间批次效应
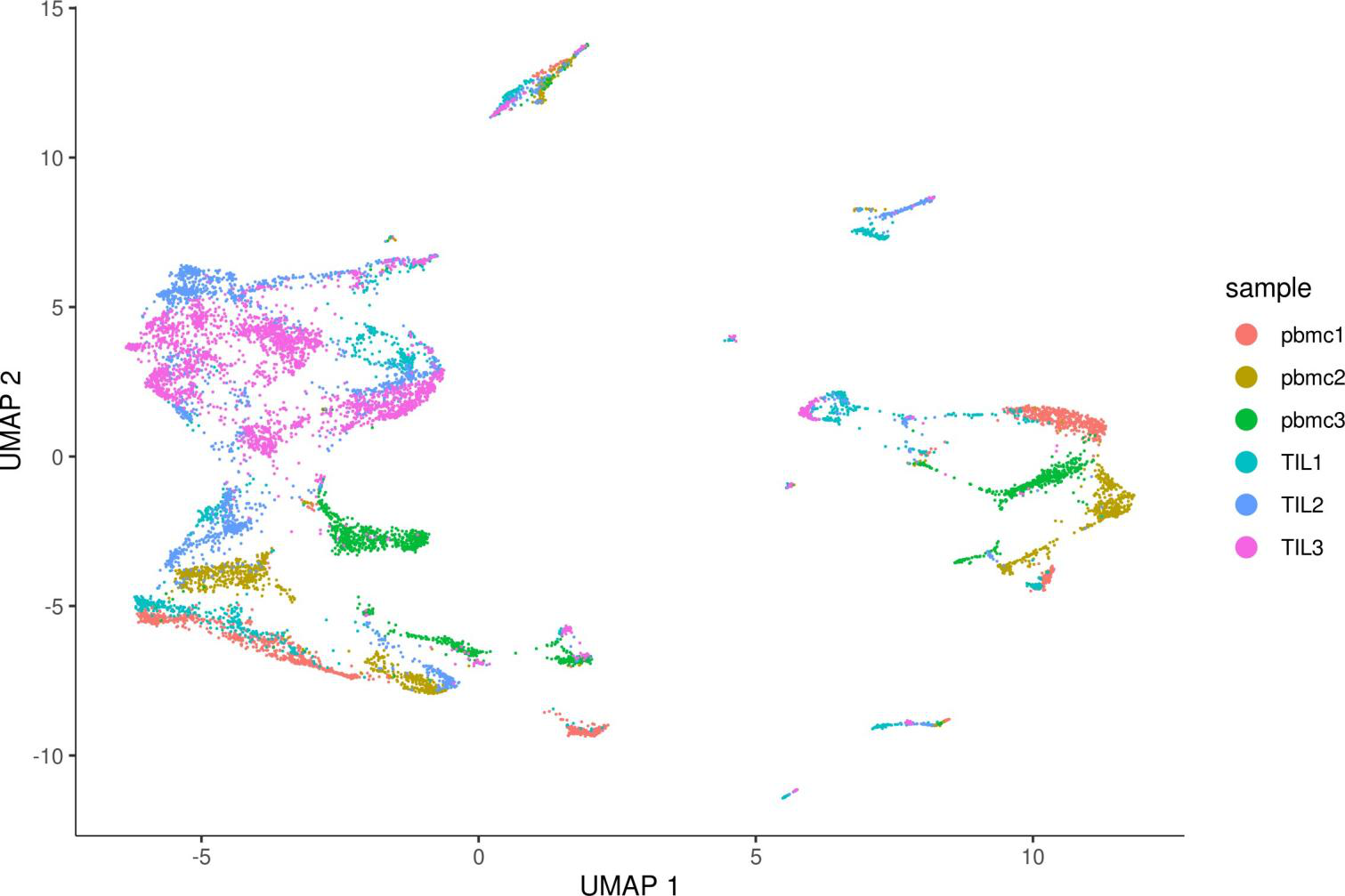
plot_cells(monocle_cds, color_cells_by=”sample”, label_cell_groups=FALSE) # 检查样本间的批次效应
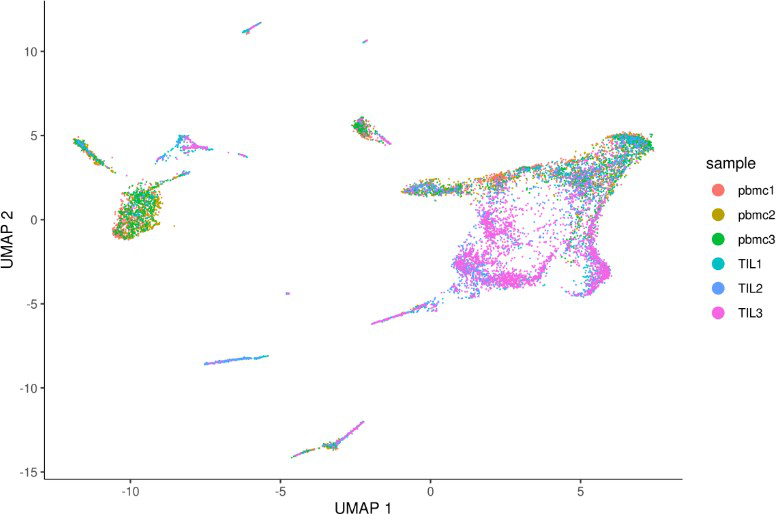
align_cds移除批次效应
1 # align_cds() tries to remove batch effects using mutual nearest neighbor alignment
2 monocle_cds <- align_cds(monocle_cds, num_dim = 100, alignment_group = “sample”) # 去除批次效应,似乎不太好使
3 monocle_cds <- reduce_dimension(monocle_cds)
plot_cells(monocle_cds, color_cells_by=”sample”, label_cell_groups=FALSE)
细胞聚类
1 # monocle3同样可以指定resolution(默认NULL),但需要注意,resolution 设置⽐ seurat⼩很多(例如当resolution=0.1时,出现⼏百个cluster)
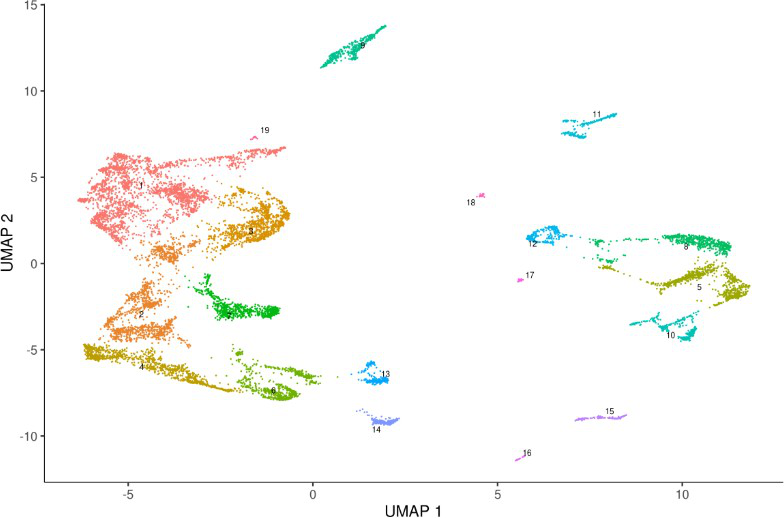
2 monocle_cds <- cluster_cells(monocle_cds, resolution = 0.0001, reduction_method = “umap”)
3 plot_cells(monocle_cds)
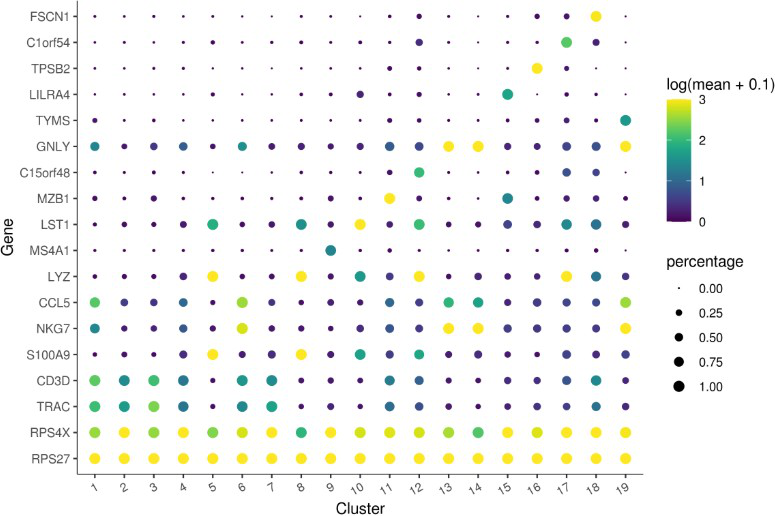
Marker 基因分析
使用 top_markers() 函数寻找cluster之间的marker基因。
1# 可以选择 cluster 也可以选择 partitions
2as.character(partitions(monocle_cds)) %>% unique
3 [1] “3” “2” “6” “11” “4” “7” “1” “5” “9” “10” “8”
4 as.character(clusters(monocle_cds)) %>% unique
5 [1] “8” “4” “10” “14” “19” “9” “15” “2” “7” “1” “6” “5” “13” “11”
“17”
6 [16] “3” “12” “18” “16”
7
8marker_test_res 9head(marker_test_res, n = 3)
10gene_id gene_short_name cell_group marker_score mean_expression 11 1 HES4 HES4 10 0.2511486 0.9178898
12 2 TNFRSF18 TNFRSF18 3 0.2701016 3.8953993
13 3 TNFRSF4 TNFRSF4 3 0.3305234 5.8852331
14 fraction_expressing specificity pseudo_R2 marker_test_p_value 15 1 0.6170213 0.4070340 0.3236830 4.873175e-105 16 2 0.7234243 0.3733654 0.2931210 9.156877e-159 17 3 0.7102540 0.4653594 0.3062058 5.322088e-166
18 marker_test_q_value 19 1 1.317746e-99
20 2 2.476093e-153
21 3 1.439135e-160
22marker_test_res 23top_specific_markers %
24filter(fraction_expressing >= 0.10) %>%
25group_by(cell_group) %>%
26top_n(1, pseudo_R2)
27top_specific_marker_ids % pull(gene_id))
28plot_genes_by_group(monocle_cds,
29top_specific_marker_ids,
30group_cells_by=”cluster”,
31ordering_type=”maximal_on_diag”,
max.size=3
 1 # 将monocle3结果存到seurat对象中
1 # 将monocle3结果存到seurat对象中
2 colData(monocle_cds)$monocle3_partitions <- as.character(partitions(monocle_cds))
3 colData(monocle_cds)$monocle3_clusters <- as.character(clusters(monocle_cds))
4 sceObj@meta.data <- pData(monocle_cds) %>% as.data.frame
百迈客不仅提供标准分析内容,还提供个性化分析内容,全新的monocle3 数据分析,详情可见单细胞转录组分析难?教您2步,轻松玩转它。更多好玩的,生信分析内容尽在百迈客云平台,如果感兴趣欢迎点击按钮联系我们,我们将免费为您设计文章思路分析方案。

推荐阅读:
SPOTlight助力单细胞和空间转录组联合分析
Cell | 单细胞+空间转录组解析人类肠道发育的时空密码
百迈客:10x单细胞转录组与空间转录组联合分析一睹为快
空间转录组ST & 单细胞转录组测序联合揭示胰腺导管腺癌的组织结构
Nature | 单细胞和空间转录组揭示类原肠胚中的体节发生
单细胞转录组+空间转录组联合绘制人类鳞状细胞癌组成和空间结构的多模式图谱
单细胞转录组+空间转录组揭示人类心脏发育过程中整个器官的空间基因表达和细胞图谱
单细胞转录组分析难?教您2步,轻松玩转它




 京公网安备 11011302003368号
京公网安备 11011302003368号