
中文名: 全球范围海洋沉积物的微生物多样性研究
英文名: Global diversity of microbial communities in marine sediment
杂志:PNAS,2020
影响因子:9.58
研究背景
海洋沉积物富含生物质,并且它是未知的微生物多样性的来源,微生物细胞密度高达每立方厘米109个细胞(比水体高出五个数量级)。海洋沉积物包括粘土,腐烂的有机物,钙质残余物和其他固体沉积物。
海洋沉积物中的微生物群落控制生态过程,影响养分供应和土壤化学特性。海洋环境是地球上最具生产力的生态系统之一,但是人为影响对于海岸生态多样性产生明显影响,导致微生物群落功能转变和人类健康风险因素。海洋沉积物中的微生物对全球生物量的贡献巨大,是地球系统的重要组成部分。海底沉积物既包括好氧微生物生态系统,也包括厌氧微生物生态系统,它们在不同的地质时期依靠着极低的生物可用能量生存。然而,海洋沉积微生物群落的分类多样性及其在全球范围内的空间分布一直没有得到深入的调查和研究。
材料方法
1、实验样本:
作者通过调查了来自全球分布的40个不同地点,共299个沉积物岩芯样品。这些样品位于海底以下0.1-678米的深度位置,样品采集后立即冷冻于-80℃环境中保存。
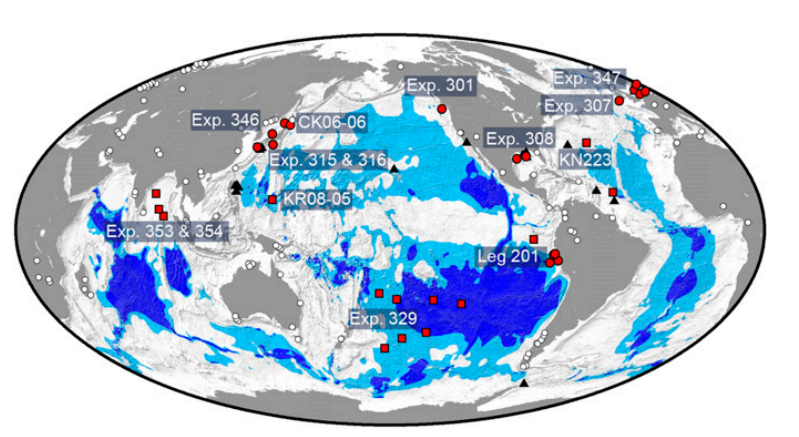
图1 采样地点的位置。红色圆圈分别表示海洋边缘站点,红色方块分别表示开放海洋站点。浅蓝色和深蓝色区域分别显示整个沉积物中可能发生溶解氧和有氧活动的最大和最小区域。在14次科学考察中,从40个地点采集了海底以下许多不同深度的299个沉积物样本,用于比较不同全球海底沉积物微生物群落组成。
2、实验和分析方法
使用试剂盒提取DNA,以提取的DNA为模板进行PCR扩增出细菌的16S核糖体区,使用引物对(U515F/U806R;518F/926R;517F/958R)进行16S rRNA基因扩增和测序,共获得超过4700万条16S rRNA基因序列。将Archaea、Bacteria和Universal datasets分别抽平至10,183、10,092和10,546条序列,并通过5个SAR模型用于鉴定多样性特征。
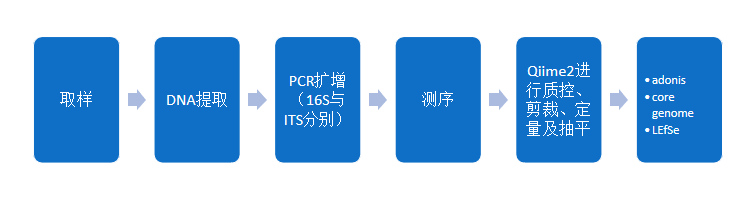
图2 实验分析流程参考
研究结果
1、古菌和细菌的群落物种组成
缺氧海底沉积物中古菌群落的分类学组成与含氧海底沉积物中的古菌群落区系组成显着不同(图3 A和C)。图3A显示了古菌测序文库的结果,用于展示235个沉积物样本的古菌群落组成,Crenarchaeota的成员分布最为广泛。Euryarchaeota和Asgardaeota在某些厌氧地区中非常丰富,例如波罗的海和Shimokita Peninsula半岛。一些来自日本海、秘鲁边缘和ShimokitaPeninsula半岛的样本中,Hadesarchaeaeota高达古菌群落的70%以上,其相对丰度随着沉积物深度的增加而增加。Nanoarchaeaeota的成员在最深层沉积物样品中丰度最高,约占比95%。而在有氧环境中,Thaumarchaeota(例如Nitrososphaera)的成员占据主导地位。在3,892条古菌扩增子序列中,在古菌数据集中检测到的ASV中有260个ASV是常见的,这260个ASV约占古菌序列总数的70%。
使用通用引物对获得的古菌群落组成与使用古菌特异性引物获得的群落组成略有不同。对于大多数沉积物样品,Asgardaeota在通用引物获得的数据集里占据主导地位,而Crenarchaeota和Euryarchaeota在古菌专用引物数据集中占主导地位。这些差异证实了引物覆盖范围会影响表观的物种群落组成。尽管两个引物扩增的数据集之间存在细微的差异,但两个数据集都表明缺氧沉积物中的古菌群落与含氧沉积物中的古菌群落显著不同。例如,古菌专用引物数据集和通用引物数据集都显示Thaumarchaeota仅在来自含氧沉积物的样品中占主导地位。
对于大多数样品而言,通用引物数据集中的古菌相对丰度低于10%(图3C),尽管由于PCR效率的差异,该结果不是定量的。先前的研究使用数字PCR定量技术来确定开放海洋沉积物和边缘海洋沉积物中古菌的百分比分别为5.6和22.6%。本研究中的百分比分别为4.3和12.2%。尽管这些百分比略低于先前的研究,但它们一致表明在海底沉积物中细菌的丰度高于古菌,且占主导地位。
与古菌群落类似,缺氧沉积物中的细菌群落与含氧沉积物中的细菌群落存在很大差异(图3B和C)。从细菌特异性引物和通用引物获得的细菌群落组成大致一致。在含氧的海底沉积物中,包括Alphaproteobacteria和Betaproteobacteriales(γ-proteobacteriaes)在内的Proteobacteria成员与Firmicutes成员一起占主导地位。相反,缺氧沉积物中普遍存在Atribacteria、Chloroflexi和Planctomycetes的成员。但是,一些缺氧沉积物,例如从孟加拉湾采集的样本,典型的含氧沉积物细菌群落占据了较高的丰度。尽管厌氧和需氧细菌群落的优势成员之间存在一致的差异,但厌氧和需氧菌群落共有30,874个ASV中的5,212个。这5212个ASV共同构成了作者数据集中16SrRNA序列的大约80%。
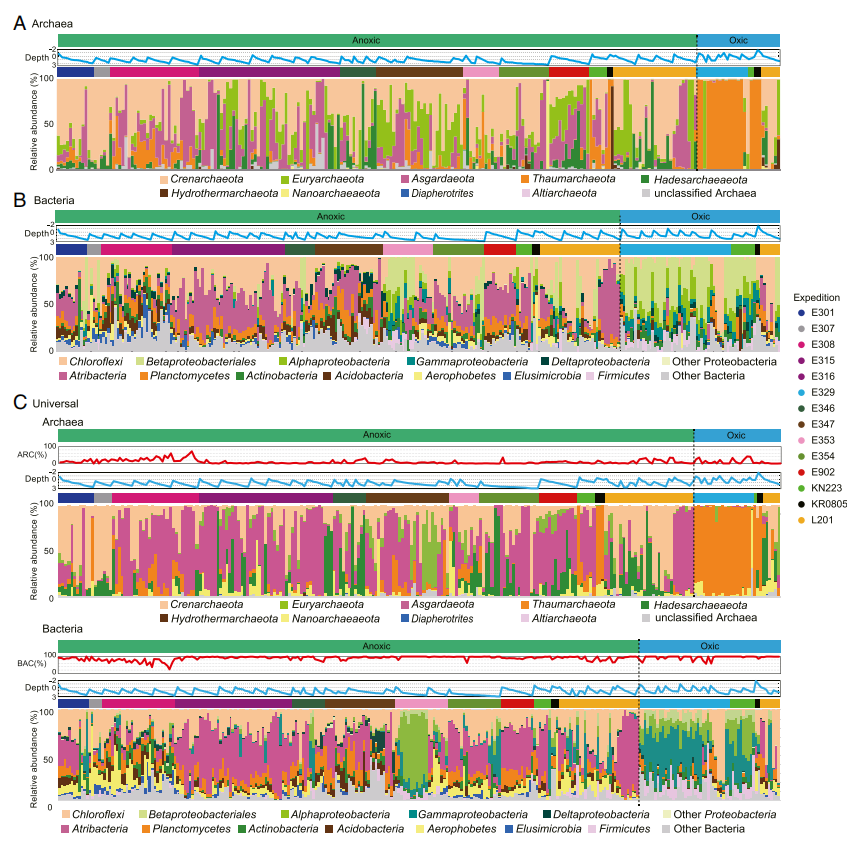
图3 海洋沉积微生物群落的物种区系组成。(A)使用古菌特异性引物获得的235个样品的古菌群落组成。(B)使用细菌特异性引物获得的272个样品的细菌群落组成。(C)使用通用引物获得的245个样品的古菌群落组成和281个样品的细菌群落组成,未显示少于1,000条序列的样本。浅蓝色折线图以对数刻度显示沉积物深度。
2、海洋沉积物中微生物群落与海水和表层土壤土微生物群落的比较
为了评估海洋沉积微生物群落与其他主要环境中微生物群落的关系,作者将海洋沉积物通用引物与使用相同通用引物的引物进行微生物群落分析的海水和表层土壤的已发表序列进行了比较。本研究中这三种生态环境的微生物群落中的细菌和古菌群落组成明显不同(图4)。如图3所示,Crenarchaeota和Asgardaeota在海洋沉积物中占优势,而Euryarchaeota和Thaumarchaeota在海水中占优势,而Thaumarchaeota在表层土壤中占优势。细菌群落组成表明变形杆菌在表层土壤和海洋中占据优势。此外,Cyanobacteria,Marinimicrobia和Bacteroidetes在海水中占主导地位,而Acidobacteria和Verrucomicrobia在表层土壤中更为重要。在海洋沉积物中普遍存在着Atribacteria和Aerophobetes,但其他两个环境中却没有这些细菌,这表明海洋沉积微生物构成了一个独特的生物群落。ASV组成的样品间相似性表明这三个生物群落中的群落不同(图4B),海洋沉积物中样品间的多样性更高。与其他环境相比,这种高多样性可能反映了海洋沉积物栖息地条件的剧烈变化。
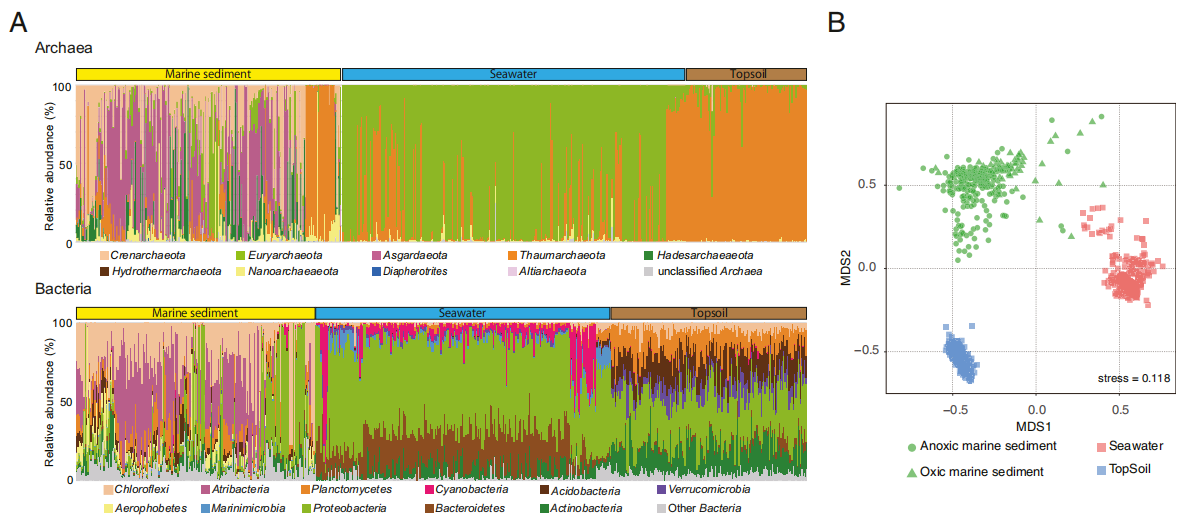
图4 比较海洋沉积物、海水和表层土壤的微生物群落组成。(A)使用16S rRNA通用引物获得的古菌和细菌群落组成。(B)使用从ASV中获得的Jaccard相似性指数值生成的NMDS排序图。
3、限制海洋沉积物中微生物群落组成的环境因素
为了了解哪些环境因素在全球范围内和广泛的沉积物深度范围内限制了微生物群落组成,作者对群落组成和各种环境特性进行了非度量多维尺度分析(NMDS)。这些环境属性包括水深、沉积物深度、硫酸盐浓度、总有机碳浓度以及是否存在溶解氧。全球范围内海洋沉积物古菌群落可分为两大类:需氧开放性海洋类和生存于富含有机物生境的厌氧类。细菌群落类似地分为有氧和厌氧的类群,它们生活在大陆边缘和其他上升流区域(图5B)。缺乏溶解氧和高浓度的有机质显着促进了海洋沉积微生物群落的物种分类组成,从而促进了好氧和厌氧海洋沉积微生物生态系统的生态功能。NMDA排序结果结果表明沉积物深度可能与群落组成独立相关。这种关系可能是由于随着沉积物深度的增加,营养物质和能量基质的可用性降低。
为了检查地理距离和沉积物岩性与微生物群落组成之间的相关性,作者对每个站点的群落组合进行了Mantel测试。地理距离与群落组成之间的正相关性相对较弱,沉积物岩性与群落组成密切相关。这种强相关性可能是由于沉积物来源和沉积速率不同而导致的不同沉积岩性中有机物的数量和组成不同所致。对于单个沉积物样品,沉积速率和沉积物年龄也可能是群落组成的重要决定因素。
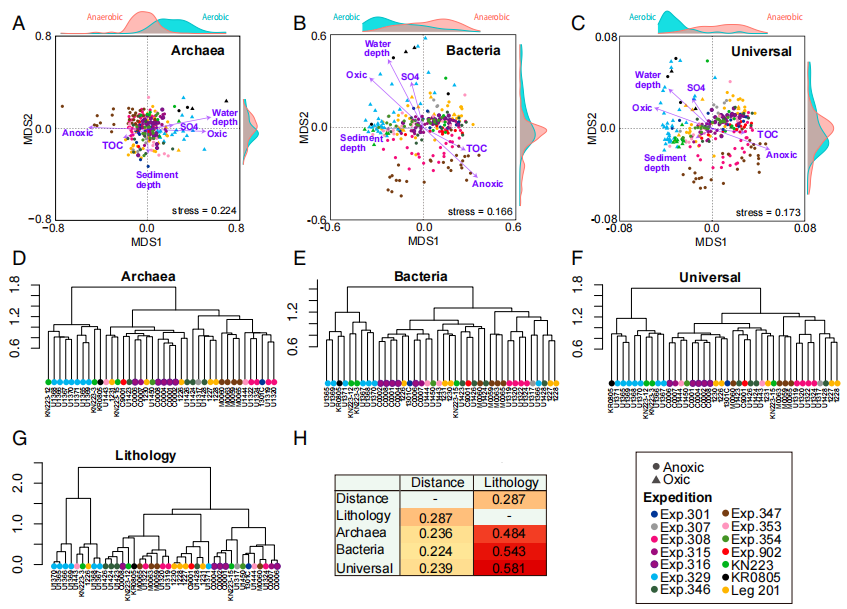
图5 海洋沉积物微生物群落的β多样性。(A–C)使用Jaccard相似性指数值生成的古菌(A),细菌(B)和通用引物数据集的NMDS排序图。紫色箭头表示与特征为P <0.01的环境因子相符的向量。(D–F)基于Jaccard相似性指数值,分别对40个地点的古菌(D),细菌(E)和通用引物数据集(F)进行算术平均(UPGMA)树状图的非加权成对分组法。(G)基于40处沉积物的矿物成分的UPGMA岩性树状图。(H)确定地理距离或岩性是否与微生物群落组成相关。
4、海洋沉积物中微生物群落组成的网络分析
为了确定共现模式,作者基于使用通用引物获得的ASV的Spearman相关性进行了网络分析(r> 0.6,P <0.05;图6)。生成的网络图包含1,533个节点(ASV),网络的模块化程度很高(0.84)。在图6B中,仅显示了10个最大的模块,其中5个群落在有氧环境中普遍存在的模块(群落1、5、8、9和10)和五个在缺氧沉积物中普遍存在的ASV组成的模块(群落2、3、4、6和7)。在这10个群落中,群落1包含最多数量的ASV(84个ASV),主要由Chloroflexi、Proteobacteria和Thaumarchaeota组成(图6C)。网络图显示,Chloroflexi和Thaumarchaeota是共现网络的重要成员(图6A)。在五个缺氧沉积物群落中,群落3主要由Atribacteria、Chloroflexi和Planctomyces组成。网络分析表明Chloroflexi是海洋沉积物环境中重要的微生物组成。但是,几乎所有Chloroflexi都与MSBL9相关联,有报道表明MSBL9适合利用多种复杂的糖聚合物。简而言之,共生网络分析表明,海洋沉积物微生物群落组成受沉积物中氧化还原态和电子供体的影响。该分析结果还表明,海底沉积微生物彼此相互作用,以有效利用这种极端环境中可用的有限基质。
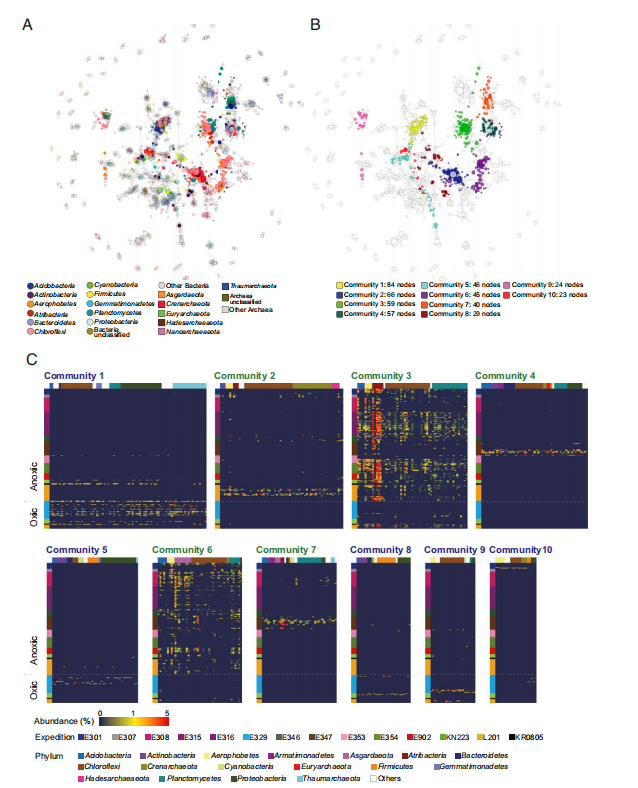
图6 共存的ASV网络分析。节点大小与PageRank成正比。
5、海洋沉积物中微生物群落丰富度的深度变化规律
为了估计每个样品的分类学丰富度,从样品的序列数据中随机抽取1,000条序列,并用于计算样本的ASV丰富度(Chao1)。细菌ASV和古菌ASV丰富度都随着缺氧沉积物深度的增加而降低(图7)。但是,在含氧沉积物中,细菌的丰富度通常随深度的增加而降低,而古菌的丰富度在整个沉积物柱中都保持相对恒定。细菌丰富度比古菌丰富度高10倍,缺氧沉积物也比含氧沉积物高(图7B),这可能是因为富含有机物的缺氧沉积物通常比缺乏有机物的含氧沉积物具有更高的微生物丰度。通常,与深度相关的丰富度下降的陡度与浅层沉积物中群落的丰富度成正相关。例如,基于缺氧沉积物中ASV丰富度和沉积物深度的回归线的斜率比含氧沉积物中的曲线陡峭(图7 A–C),与含氧沉积物相比,无氧沉积物中较小的初始丰富度在海底选择压力下得以生存。类似地,在缺氧和含氧沉积物中,随着沉积物深度的增加,古菌的富度下降不如细菌富度大。古菌丰富度随深度降低的程度较低,这与古菌类群比细菌类群对低能量可利用性的一般耐受性有关。然而,尽管古菌丰富度随沉积物深度降低的速率较低,但在所有沉积物深度,古菌丰富度通常都低于细菌丰富度。
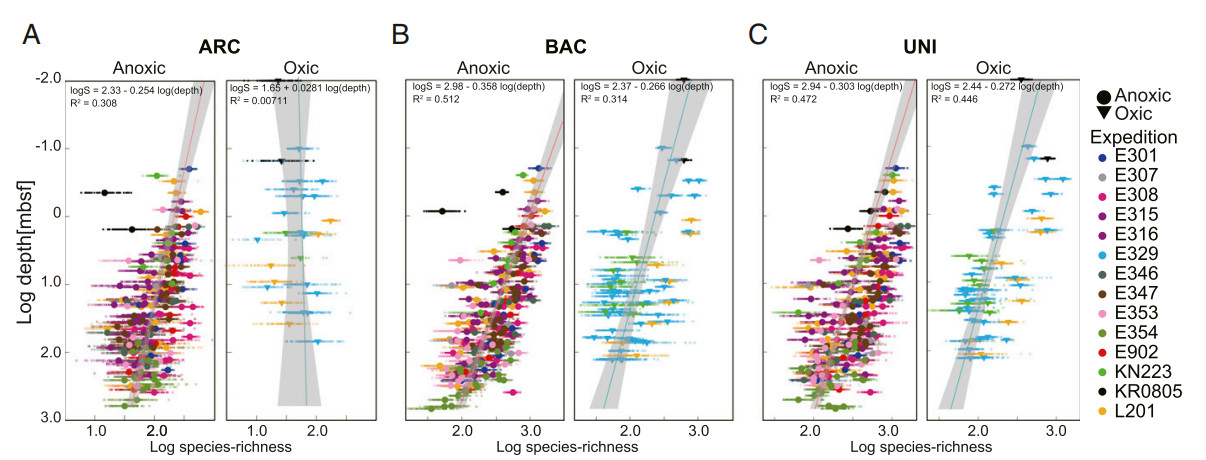
图7 含氧海洋沉积物和缺氧海洋沉积物中微生物丰富度的深度分布。
6、海洋沉积物中微生物群落的总分类学丰富度
作者使用了三组16SrRNA基因序列文库(古菌,细菌和Universal)来估计海洋沉积物中ASV的数量。为了确保作者的结果的可信性,作者同时利用以上五种SAR方法估计了其他生境中古菌和细菌多样性,包括表层土(topsoil)、海水(seawater)、海底(subseafloor)和海洋(ocean),结果可作为参考数据使用,发现测序深度和是否去除污染物对使用SAR方法估计多样性差别不大。
在这五个模型中, AIC得分表明,asymptotic模型和Gitay模型对于这三个数据集而言是最佳和次佳的。如果将其外推到海洋沉积物的总量,则使用asymptotic模型时古菌ASV的总数为7.85×103,而使用Gitay模型时则为6.10×105。使用asymptotic模型,全球海洋沉积物中细菌ASV的总数为3.28×104,而使用Gitay模型,则为2.46×106。在这两个模型中,细菌丰富度大约比古菌丰富度高四倍。对于古菌,模型外推的范围是6.18×103至7.90×1015,对于细菌ASV,模型外推的范围是2.88×104至1.32×1015。使用通用数据集估算全球多样性,作者再次发现asymptotic模型是最佳模型,古菌和细菌的多样性分别为1.98×103和3.90×104。该结果与上述细菌和古菌数据集的结果一致。使用asymptotic模型和Gitay模型进行的估算远低于先前对地下地下生物群落中微生物分类学丰富度的估算(即109至1012)。作者认为使用Arrhenius模型计算的全局ASV值是一个很大的高估。
总结
在本研究中,作者调查了来自全球分布的40个不同地点,共299个沉积物岩芯样品。这些样品位于海底以下0.1-678米的深度位置,通过高通量测序共获得超过4700万条16S rRNA基因序列,较大的测序深度使得所有样本之间能够互相进行准确的比较。
统计分析表明,物种分类组成、沉积有机碳浓度和溶解氧之间存在显著的相关性。物种-区域拟合关系模型发现海洋沉积物中古菌和细菌的物种丰富度分别为7.85×103~6.10×105和3.28×104~2.46×106,表明海洋沉积物中微生物的丰度大小可以与表层土壤和海水的物种丰富度相媲美。




 京公网安备 11011302003368号
京公网安备 11011302003368号