
今天给大家带来一篇新鲜出炉的多组学联合分析文章的案例解读,这篇文章也是北京百迈客公司和浙江大学合作的一篇成功案例。本篇文章主要利用了DNA甲基化、转录组、代谢组进行了多组学联合分析,阐述了相近地域但是土壤生态条件差异巨大的两个野生大麦种群的物种形成机理和生态逆境适应性机制。
北京百迈客生物科技提供基因组、转录组、代谢组等多组学的测序分析服务,针对性的设计项目执行方案,百迈客云平台搭载多组学联合分析平台,助力您的深度个性化挖掘!
英文题目:Multi-Omics Analysis Reveals the Mechanism Underlying the Edaphic Adaptation in Wild Barley at Evolution Slope(Tabigha)
发表期刊:Advanced Science
影响因子:16.803
研究背景
土壤是植物的活基质,为植物的生长发育、水、 矿物元素和微生物群提供了必要的物理支持,土壤的物理、化学和生物特性对植物的生长发育有很大的影响。因此,通过开发适合各种类型土壤的更好的作物品种,可以实现土壤资源的可持续和有效的利用。大麦一直是一种重要的粮食 作物,现在也被用作饮料和饲料的原料生产产品。在大麦驯化过程中,特别是在现代育种和集中化栽培后,栽培大麦的遗传多样性显著降低,成为大麦育种提高生物非生物压力耐受性和改良的瓶颈,野生大麦已适应各种土壤 类型、有效性、水分、温度和海拔高度不同的环境,成为改良栽培大麦的理想遗传资源。已有研究已发现了多个物种(包括病毒、细菌、真菌、植物和动物)对微生态环境的适应性及产生的遗传变异。当植物暴露于环境胁迫下时,DNA甲基化对转录组的强调控已被广泛报道。植物对环境胁迫的反应是一个复杂的过程,需要在多组学水平上进行协调,目前基因组测序、甲基化组测序、转录组,蛋白质组学,和代谢组在这方面都有了成熟的应用。水稻、苹果等物种的非生物胁迫研究表明植物中DNA的甲基化和TE的动员可以影响适应性应激反应和基因组的稳定性,因此,破译小麦等野生品系的DNA甲基组与转录组和代谢组的相互作用,将有利于耐非生物和生物胁迫新品种的选择和育种。
实验材料
巴勒斯坦(Tabigha)的 “进化坡”上的两个野生大麦种群Basalt和Terra Rossa的实验室水培材料,两个品种各取5份三叶期17日龄幼苗进行DNA甲基化(WGBS)测序,并分别对每份材料的根、叶组织分别设置3次重复进行转录组及代谢组的测序。(其中DNA甲基化、转录组测序分析由北京百迈客提供支持)
结果分析
一、大麦种群Basalt和Terra Rossa的甲基化分析
通过WGBS测序技术在10份野生大麦材料中共获得1620Gb的cleandata,共鉴定出131408470个CG甲基化位点(mCG)(占所有CGs的94.7%)、 97714324个mCHGs位点(占所有CHGs的77.6%)和20576899个mCHHs位点(占所有CHHs的2.9%)。并且一共鉴定出121433个差异甲基化区域(DMRs),其中52306个CG-DMRs,30720个CHG-DMRs和38407个CHH-DMRs(图C)。通过甲基化区域在染色体上的分布,作者发现CG-DMRs和CHH-DMRs两种类型的差异甲基化区域在染色体的远端区域高度积累,并且主要集中在基因区和启动子区,而CHG-DMRs的情况则和它们不同,不仅在染色体上分布相对均匀,并且基因区和启动子区的表达也相对比较低(图A)。这些结果表明,CG-DMRs和CHH-DMRs可能在野生大麦两个土壤种群的适应性功能多样化和基因区域调控中发挥重要作用。对CG-DMR基因和CHH-DMR基因进行GO富集分析,发现CG-DMR主要富集在与基本过程相关的基因中,如ATP的产生、转录和翻译,而CHH-DMR基因在代谢过程中富集,包括碳代谢、糖代谢和应激反应途径(图H)。
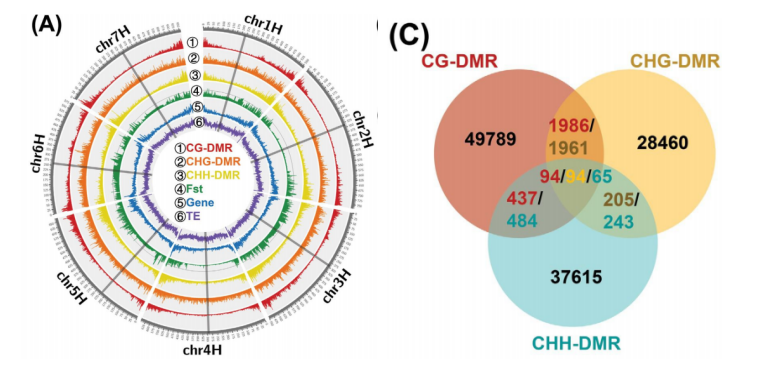
(A) 基因组中甲基化区域、Fst、基因和转座子元件(TE) 的分布密度 (C) 三种DMR类型的维恩
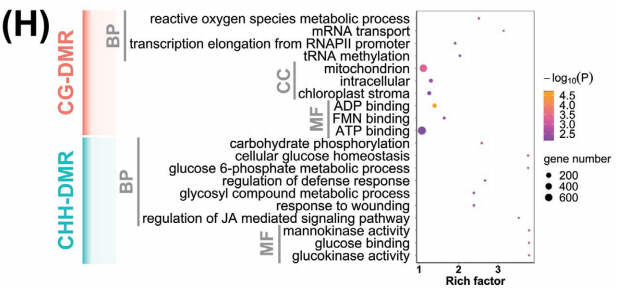
DMR相关基因的GO富集
二、. 两种野生大麦的转录组及代谢组分析
通过转录粗测序技术,共获得(431.97Gb)的cleandata。与Basalt大麦种群相比,Terra Rossa大麦种群在叶片和根系中分别有712和567个上调表达基因,497和752个下调表达基因(图A,C)。根据KEGG富集分析,差异基因在叶片和根中的“苯丙烷生物合成”、“植物-病原体相互作用”和“淀粉和蔗糖代谢”代谢途径中均富集(图2E,F),但是“苯丙烷生物合成”、“植物-病原体相互作用”途径的相关基因在Basalt大麦种群中的表达更高,而“淀粉和蔗糖代谢”途径的相关基因则Terra Rossa大麦种群中表达更活跃(图B,D)。
代谢组是生命活动的直接体现者,作者利用利用UPLC-Q- TOF/MS对两个野生大麦土壤种群的代谢物谱进行了分析,在叶片和根中分别鉴定出157种和147种代谢物,Terra Rossa大麦种群叶片中糖、三羧糖循环中的有机酸和氨基酸的浓度较高,这与材料中水解酶基因较高的转录活性相一致。而Basalt大麦种群的叶片和根均表现出较高浓度的酚酰胺及其衍生物(图2G、H)(主要与更活跃的苯丙烷 途径相关)。此外,在Basalt大麦种群根中观察到谷胱甘肽的富集(主要与GSTs的高表达有关)。综上所述,转录组和代谢组均表明,Terra Rossa大麦种群在初级代谢中活性较高,而Basalt大麦种群材料的次生代谢活性更活跃。
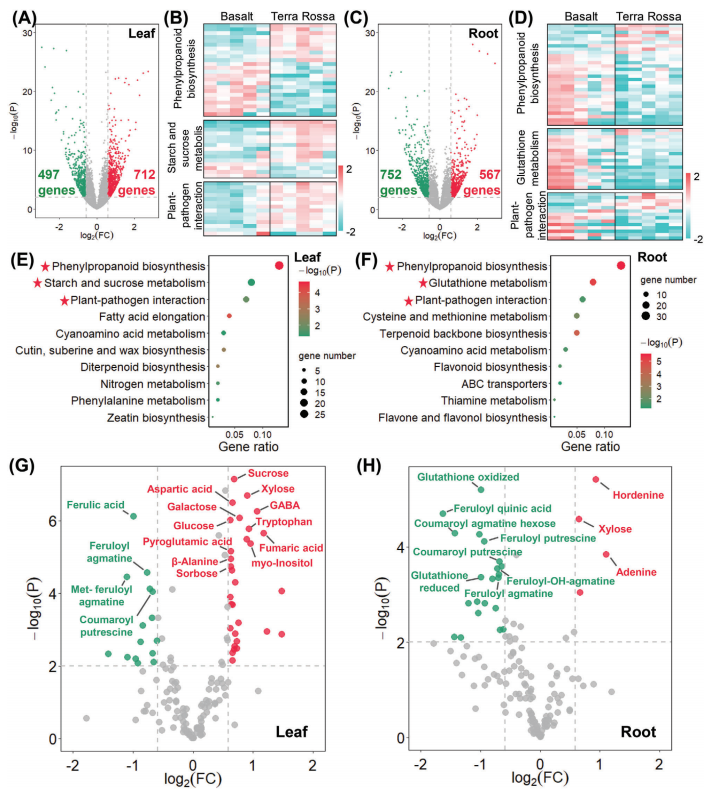
(A、C)叶片和根中差异表达基因(DEGs)的火山图。(B、D)deg的热图。E,F)叶和根中DEGs的KEGG富集。(E,F)叶和根中DEGs的KEGG富集。(G,H)代谢物水平倍数变化的火山图。
三、DNA甲基化对mRNA和lncRNA的表达调控
很多研究表明,DNA甲基化在基因表达的调控中起着至关重要的作用。靠近TSS的上游区域的CG和CHG甲基化水平急剧下降,且上游100bp的甲基化水平与基因表达水平呈负相关(图B),而CHH甲基化水平却呈现先上升再下降得趋势,并且在 1000~200bp的上游区域,CHH甲基化水平与基因表达水平呈正相关(图C)。为了确定高或低甲基化对单个基因表达的影响,作者对10份野生大麦品种的甲基化水平与基因以及lncRNA表达水平进行了Pearson相关分析,发现对于部分基因,近转录起始位点启动子区域的CG和CHG甲基化会抑制mRNA转录,而CHH甲基化则会促进mRNA的表达。
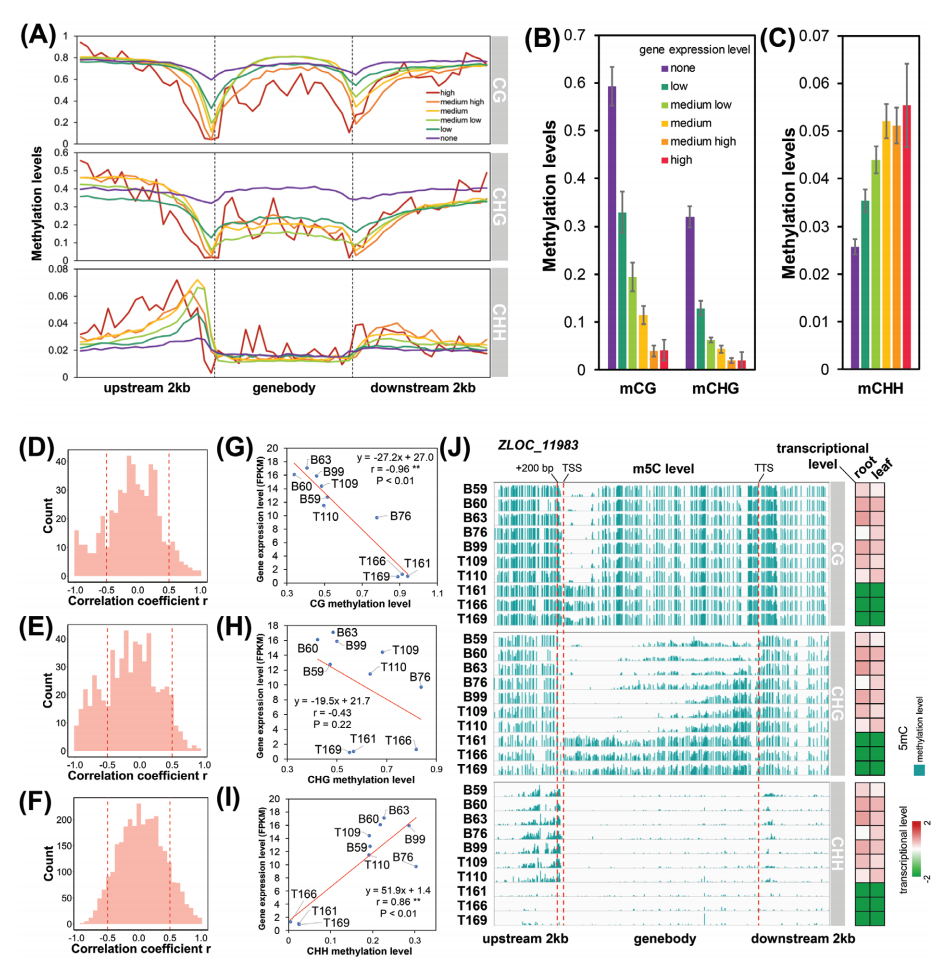
(A)被不同表达水平划分的基因内和侧翼的甲基化水平。(B)TSS上游100bp的平均CG和CHG甲基化水平。(C)平均CHH甲基化水平TSS上游1000~200bp的水平。(D-F)m5C水平(D、mCG、E、mCHG、F、mCHH)与转录水平之间的Pearson相关系数分布。(G-I)m5C水平(G、mCG、H、 mCHG、I、mCHH)与ZLOC_11983转录水平的相关性分析。(J)m5C水平和ZLOC_11983的转录水平。
结论
本研究通过DNA甲基化分析,发现表观基因组和基因组变异导致了进化坡(Tabigha)的两个野生大麦毗邻的地质和土壤种群之间的差异。此外,通过代谢组和转录组的研究,发现糖代谢的促进有助于野生大麦对进化坡(Tabigha)Terra Rossa土壤的土壤适应,而苯丙烷/苯酰胺生物合成和免疫信号通路的增强使野生大麦能够后期适应进化坡(Tabigha)的潮湿、富含真菌和细菌的Basalt土壤。总之,野生大麦在对比土壤的土壤适应性可能在表观基因组、转录组和代谢组水平上都受到调控。另外,研究中发现的关键基因以及抗真菌代谢物可以作为提高栽培大麦和其他谷类作物非生物和生物耐受性的重要遗传资源,以确保粮食可持续生产和粮食安全。
产品介绍
转录组测序(RNA-seq)能够对样品任意时间点或任意条件下的转录组进行测序,获取某一状态下所能转录出来的所有mRNA的信息,拥有√确到单个核苷酸的分辨率。能够动态反映基因转录水平,同时鉴定和定量稀有转录本和正常转录本,并且提供样品特异的转录本序列结构信息。目前已经广泛的应用于动植物发育调控、环境适应、免疫互作、基因定位、物种遗传进化及肿瘤与遗传病检测等。
代谢组学(meta-bolomics)是对不同生物体、组织或细胞所有小分子代谢物进行分离、定性和定量分析,并寻找代谢物变化规律或差异与生理、病理、表型变化的相对关系,进而解析生物学问题。




 京公网安备 11011302003368号
京公网安备 11011302003368号