

文章题目:Utilizing PacBio Iso-Seq for Novel Transcript and Gene Discovery of Abiotic Stress Responses in Oryza sativa L.
中文题目:利用Pacbio Iso-Seq测序技术发现水稻非生物胁迫下的新转录本和新基因
发表期刊:International Journal of Molecular Sciences
发表时间:2020年10月31日
影响因子:4.556
研究背景
全球气候变化导致高温、干旱和夜间高温等非生物胁迫条件的严重程度和频率增加,这些都造成了作物产量的降低。随着世界人口的增长,植物育种专家面临着开发高产、优质、减少环境污染的新品种的艰巨任务。水稻是世界上一半以上人口的主要卡路里来源,特别是对亚洲最贫穷的人来说。世界各地的基因库中保存着23万多份水稻及其野生近缘种的广泛自然遗传多样性的种质资源,是一种无价的可用于作物改良的重要基因库。
虽然世界上近80%的水稻种植是基于indica(籼稻亚种)品种,但目前的标准基因组及其注释来自粳稻亚种Nipponbare。由于缺乏适当的基因组,不同水稻亚种的研究大多都基于Nipponbare基因组。例如,在3000水稻基因组计划中将测序序列比对到Nipponbare基因组上,丢弃了不能比对到该参考基因组的序列。这可能会导致非粳稻亚种特有的遗传信息的丢失。另外,最近已经对其它水稻亚种的栽培品种的基因组进行了测序,例如indica(Shuhui498,Zhenshan 97, Minghui 63)、aus(Kasalath,N22),但其完整性和注释程度仍存在差异。值得注意的是aus亚种是抗病、耐磷酸盐缺失、耐涝、耐厌氧发育和抗旱等潜在性状的宝贵基因来源。例如,在aus品种基因组中发现了耐磷酸盐缺失相关基因OsPSTOL1、耐涝相关基因OsSNORKEL1/2和OsSUB1A。值得注意的是,这些基因在粳稻的Nipponbare亚种基因组序列中是不存在的。
在过去的几年里,RNA测序(特别是基于illumina的短序列RNA-seq)已经成为分析转录组的有力工具,用来识别在非胁迫控制和各种环境胁迫条件下差异表达的基因。然而,需要基于参考基因组或转录组序列对RNA-seq数据进行比对和注释来确定转录水平。在水稻中,参考基因组决定了可以鉴别的差异表达基因和转录本亚型。显然,参考基因组/转录组中没有的基因的表达信息在分析过程中会丢失。这在研究耐胁迫的外来品种、陆地品种或野生稻种时尤其相关,因为它们可能含有参考品种Nipponbare不存在的耐受基因。这将严重限制识别支持作物改良计划的新候选基因的可能性。
解决这一问题的一个显而易见的办法是对所需的基因组进行测序、组装和注释。但是,这种方法比较昂贵和耗时。在这篇文章里,我们探索了一种更有针对性的RNA-Seq序列方法来测序和重建了三个不同亚种的水稻品种的部分转录本作为参考,Pacific Bioscience(PacBio)属于提供高通量全长转录本序列的新一代测序方法。该方法已成功应用于对现有植物转录本及注释的探索和扩展,如高粱、小麦、甘蔗、野生棉花、不同的穗型草、苜蓿等。
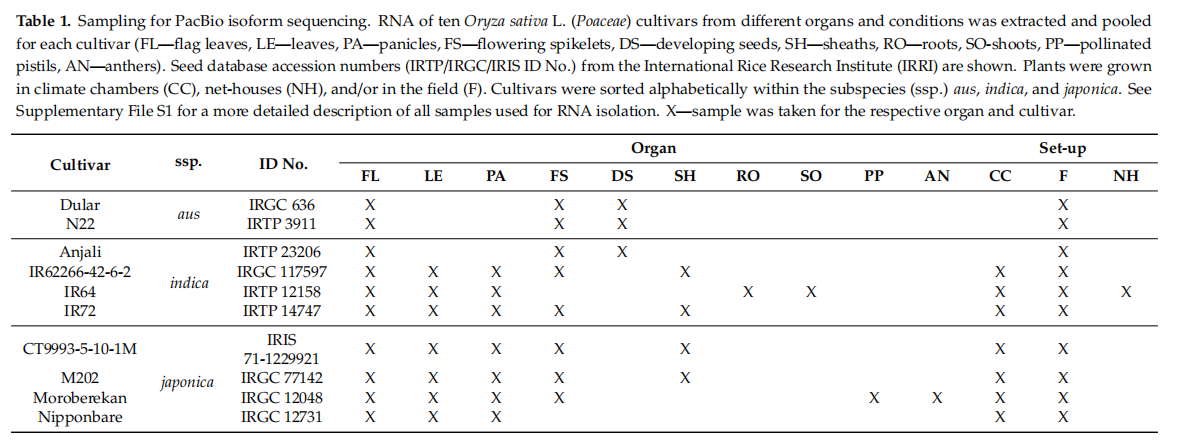
取样材料
样品取自三个水稻品种的10个不同亚种的不同组织部位(表1):
分析结果
1.重构转录本
使用PacBio Sequel I测序平台对每个品种进行SMRT测序,得到15.49~24.51GB的转录本数据。用IsoSeq3软件对原始测序数据进行ccs和lima处理,每个品种SMRT cell分别得到460340~736747条全长非嵌合序列(full-length non-chimeric reads简称FLNC,包含3 ‘ 引物、5 ‘ 引物以及polyA尾)。全长非嵌合经过IsoSeq3聚类和polish分别得到37951~54684高质量转录本(HQ)以及1233 ~2170低质量转录本(LQ)。先将HQ与NCBI核苷酸数据库进行blastn比对(E<=1e-10),再将上一步为比对上的转录本序列与NCBI蛋白数据库进行blastx比对(E<=1e-10),去除未比对上两个数据库的转录本序列,最终得到37535~54594条HQ用于后续分析(表2)。
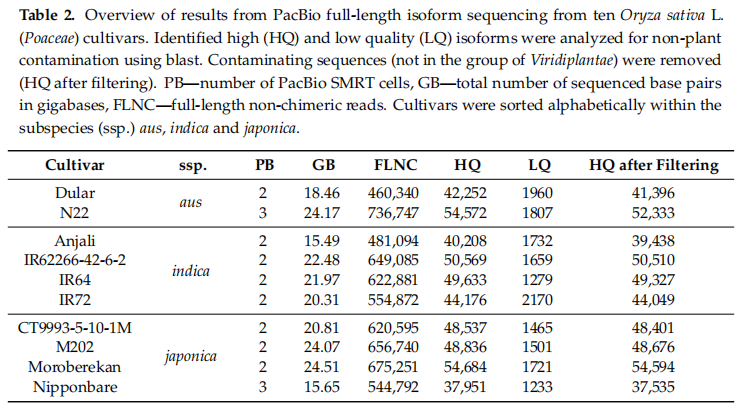
Pacbio RSII平台声明使用RNA-seq二代测序数据对转录本数据进行矫正,可以得到更多的HQ序列,因为LQ序列中含有大量的插入和缺失。然而与RSII相比,PacBio Sequel I测序平台的测序结果更好。为了验证这一结果,我们用minimap2将未矫正的HQ比对到相应亚种的基因组,结果表明缺失的比例很小,所以进一步的分析中没有包含LQ序列。
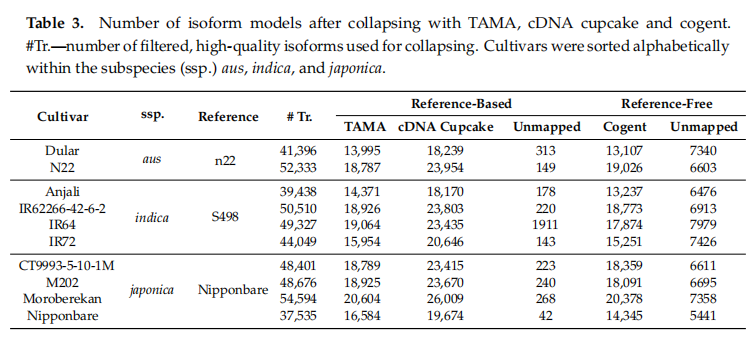
2.转录本去冗余
在文库准备过程中,会产生5 ‘ RNA降解产物,并进行测序。这些降解产物具有相同的外显子结构,但缺乏5 ‘序列信息,因此产生与技术偏差或生物学背景无关的冗余异构体。我们测试了三种不同的去冗余方法,包括cogent、cDNA cupcake和TAMA,其中cDNA cupcake和TAMA需要基于参考基因组,而cogent不需要基于参考基因组。cogent基于pacbio全长转录本序列重构一个参考基因组,然后将相同的序列比对到重建的基因组,基于比对结果利用cDNA cupcake算法对转录本去冗余。cDNA cupcake和TAMA直接用minimap2软件和各自的亚种参考基因组进行比对。基于这三种方法,只有很少的转录本不能回比到基因组上(表3)。总的来说,这三种去冗余方法均能显著减少异构体的数量,分别为47.6% (cDNA cupcake,Nipponbare)和68.3%(cogent,Dular)。
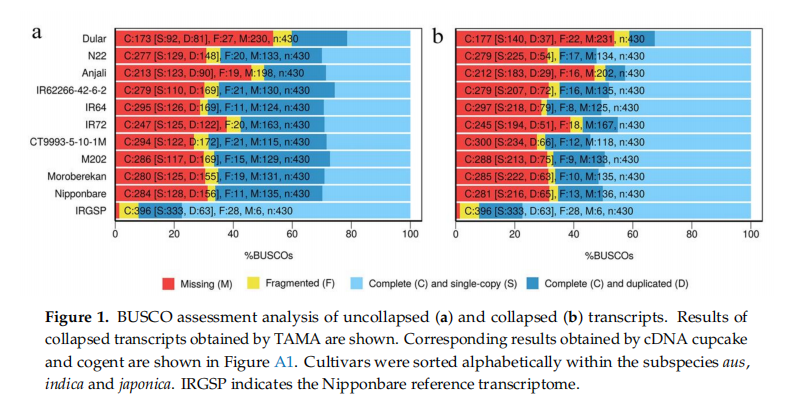
基于植物中430个高度保守的同源蛋白利用BUSCO软件对TAMA算法去冗余前后的HQ进行完整性评估(图一),由于取样不完全,缺失了54%~27%的重要蛋白,其中Nipponbare参考基因组(IRGSP)只缺失了6种。cDNA cupcake和TAMA的结果相似,而对于cogent,超过50%的蛋白缺失,最有可能的原因是转录本没有回比到重建的基因组。
去冗余后转录本长度中值都有所增长,长度分布和转录本长度中值与Nipponbare参考基因组相似。统计了去冗余后10个品种每个基因相应的转录本数量,其中基因只有一个转录本的比例,TAMA最高达到了75%,cDNA cupcake在60%左右,cogent只有50%。同时计算了Nipponbare参考基因组每个基因对应的转录本数量进行比较,该参考基因中基因只有一个转录本的比例达到了85%(图2)。
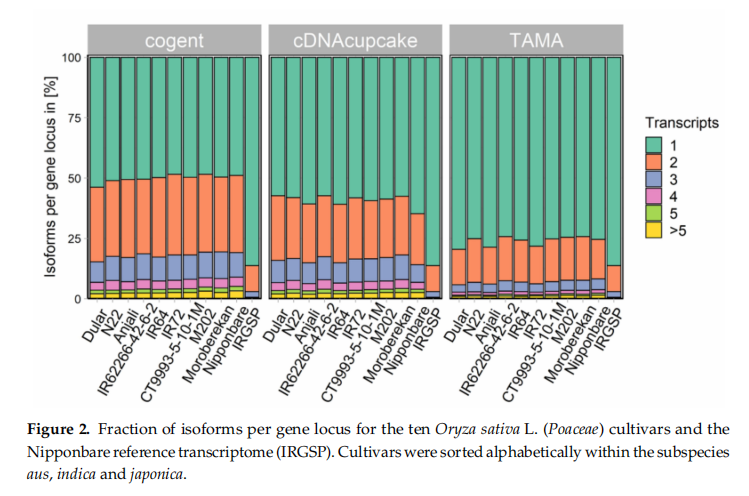
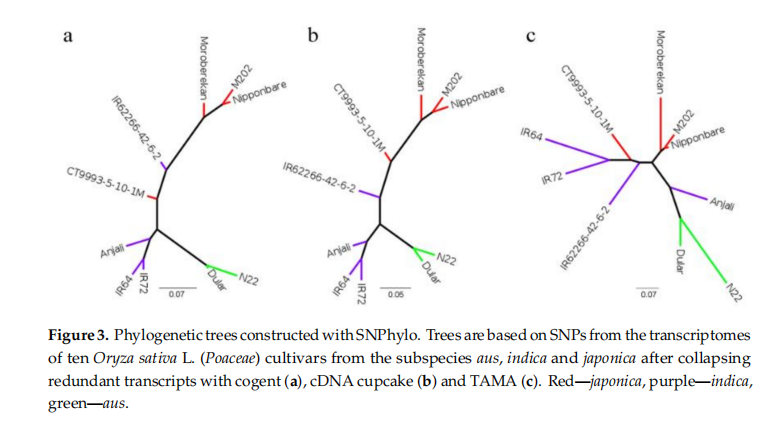
来自同一亚种的不同品种亲缘关系更近,我们使用系统发育树评估亚种之间的遗传距离。利用去冗余后的转录本序列基于Nipponbare参考基因组识别SNPs,使用SNPhylo绘制进化树(图3)。SNPhylo提取高质量并且具有代表性的SNPs进行后续分析,cDNA cupcake算法大约30000个SNPs,cogent算法大约23200个SNPs,TAMA算法大约16000个SNPs。三种方法中,同一亚种的不同品种聚类在了一起,cDNA cupcake算法和cogent的聚类结果更相似。三种方法均能将aus与另外两个亚种区分开,但cogent和TAMA对indica和japonica种间的区分不如cDNA cupcake明显。
3.评估重构的转录本
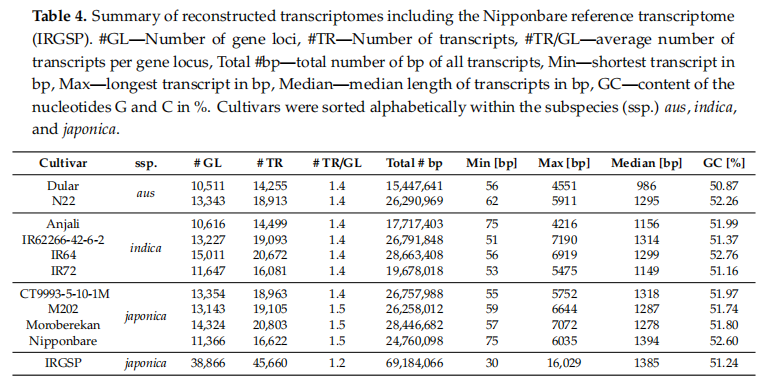
基于TAMA算法得到的HQ进行转录本的评估。由于TAMA只对比对到参考基因组上的转录本进行去冗余,我们用cogent对没比对上参考基因组的转录本进行去冗余。合并结果后,每个品种最终得到10511(Dular)~15011(IR64)个基因,14255(Dular)~20803(Moroberekan)个转录本(表4)。与Nipponbare参考基因组相比,大约三分之一的基因位点和大约一半的转录模型被重建。每个品种每个基因的平均转录数约为1.4~1.5,略高于参考基因组的1.2。中位转录本长度为986 bp(Dular)~1394 bp(Nipponbare),与Nipponbare参考值1385 bp相似。平均GC含量在50.87%(Dular)~52.76%(IR64),与Nipponbare参考值51.24%相似。利用gffcompare软件与Nipponbare参考基因组进行比较识别新基因与转录本。
4.功能注释
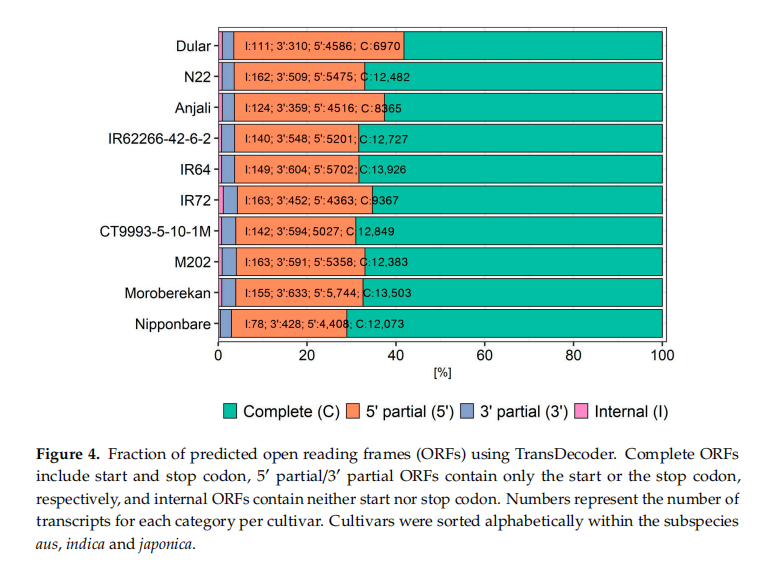
为了深入了解重建转录本的生物学信息,我们进行了功能注释。使用TransDecoder软件预测开放阅读框(ORFs),包括blast和PFAM,结果表明大约有60%~70%的完整ORFs(包括启动和终止密码子)。此外还发现了26%~38%的5 ‘ ORF、很少比例的3 ‘ ORF和中间ORF(既没有起始密码子也没有终止密码子)(图4)。
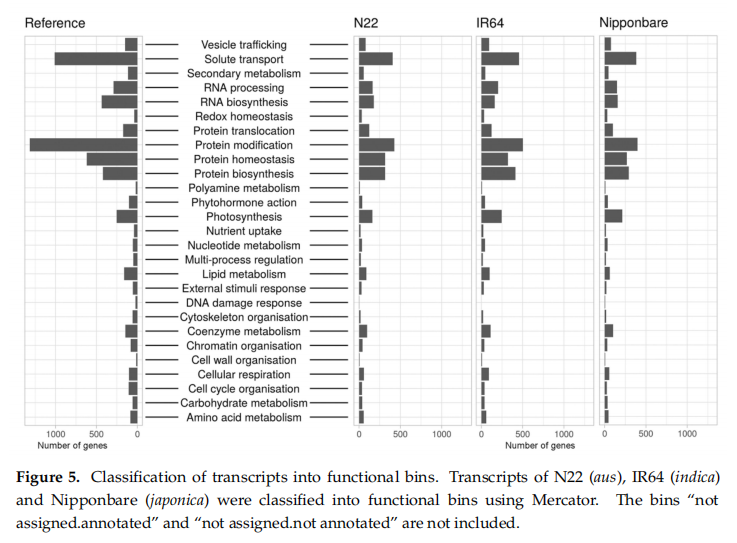
使用Trinotate和Mercator4进行功能注释。Mercator4是专门为植物开发的,它使用了一种简单的层次树结构,被称为“容器”,用来描述生物学概念。主要的生物过程如光合作用,都是由顶层的容器来表示的,每个子容器描述的是一个更详细的子过程。目前本体包括27个功能类别,代表了植物中不同的生物过程。N22、IR64和Nipponbare三个品种作为各自亚种的代表与植物中所有水稻基因的分类进行比较分析,结果显示三个品种的注释结果分布相似(图5)。超过28000个水稻已知基因在Mercator库中没有注释分类信息,因此三个品种有8000~10000个转录本没有分类注释到Mercator库。
5.品种间共有和特有的转录本
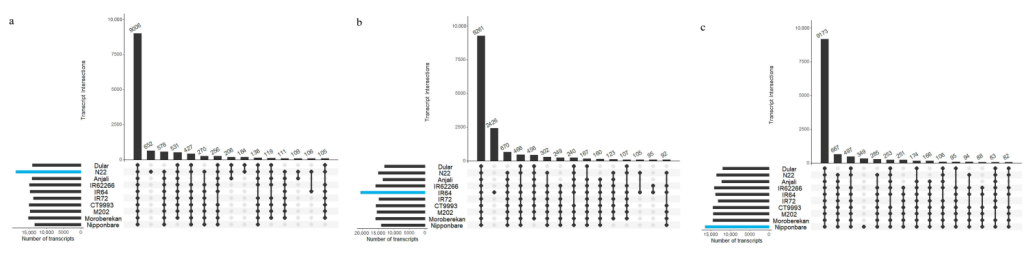
为了鉴定品种特异性转录本,以N22、IR64和Nipponbare三个品种的转录本作为blast比对库,其它9个品种与其进行比对(图6)。识别到N22特有转录本652个,IR64特有转录本2426个,Nipponbare特有转录本349个。

6.aus特有转录本的差异表达分析
aus品种N22特别抗旱和耐热胁迫,因此我们想知道在这些条件下是否有aus特异转录本受到调控。以N22为研究对象,分析干旱和热胁迫下差异表达的基因。利用从发育种子中分离的RNA进行Illumina测序,将测序数据回比到重构的N22转录本。使用DESeq2基于参数FDR<0.1和|log2FC|>=1软件识别出56个aus特异的差异表达基因。这56个差异基因进行blast比对,其中46%比对上拟南芥,27%没有任何注释信息,11%仅描述了一个PFAM域或与其它植物物种的序列同源,而在水稻中仅有5%已知同源基因。
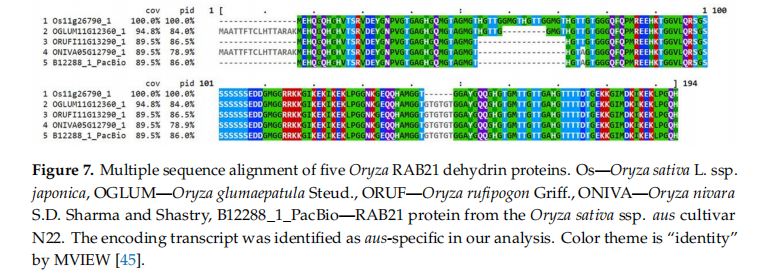
举个例子,在高温和干旱双重胁迫下显著上调的基因B12288。它在japonica和indica中均有同源基因RAB21,这个基因受干旱的诱导,其编码的蛋白属于LEA脱氢蛋白家族。与水稻其它脱氢蛋白进行多序列比对研究(图7),N22实际上的基因与野生稻、O. sativa ssp. japonica中其它4种脱氢酶的亲缘关系密切。序列覆盖率为89.5%,序列同源性86.0%,其中包含脱氢酶高度保守的重复区。相比japonica蛋白,N22蛋白序列与野生稻更接近。
总结
本文主要探讨了Pacbio Iso-Seq获得的转录本相比于Nipponbare参考基因组是否可以用于aus等水稻亚种的下游分析。此外通过这些转录本,我们希望发现水稻非生物胁迫下新的转录本和基因。我们的分析表明所有品种都可以鉴定出特异的转录本,还确定了aus亚种特异的差异表达基因。Pacbio Iso-Seq这种方法适用于其它没有基因组或者基因组质量不高的物种,相比对基因组组装,这种方法更省时便宜。




 京公网安备 11011302003368号
京公网安备 11011302003368号