
简介
单细胞分析之细胞间通讯分析能够帮助我们解读细胞与细胞之间的相互作用。目前单细胞通讯分析的工具有很多,小编在这里主要介绍 celltalker。celltalker根据 ligand 和receptor 的表达,细胞 cluster 以及样本分组来评估cell-cell communication。
celltalker主要功能函数介绍
reshape_matrices():Reshape matrices,重构数据,为每个样本分配表达矩阵
create_lig_rec_tib():Create a tibble of consistently expressed ligands and receptors,为每个分组创建一致的配体-受体表达
putative_interactions():Create a tibble of consistently expressed ligands and receptors,推断 ligand-receptor pairs
unique_interactions():Unique interactions between two groups,识别分组中唯一的互作
circos_plot():Creates a circos plot from the list of ligands and receptors,绘图
读入单细胞数据
数据来源:来自 GSM4138110 的6个样本:分别是:HNC_1_PBMC、HNC_2_PBMC、HNC_3_PBMC、HNC_1_TIL、HNC_2_TIL、HNC_3_TIL
sce.ob <- readRDS(“F:/single_seruat.Rds”)
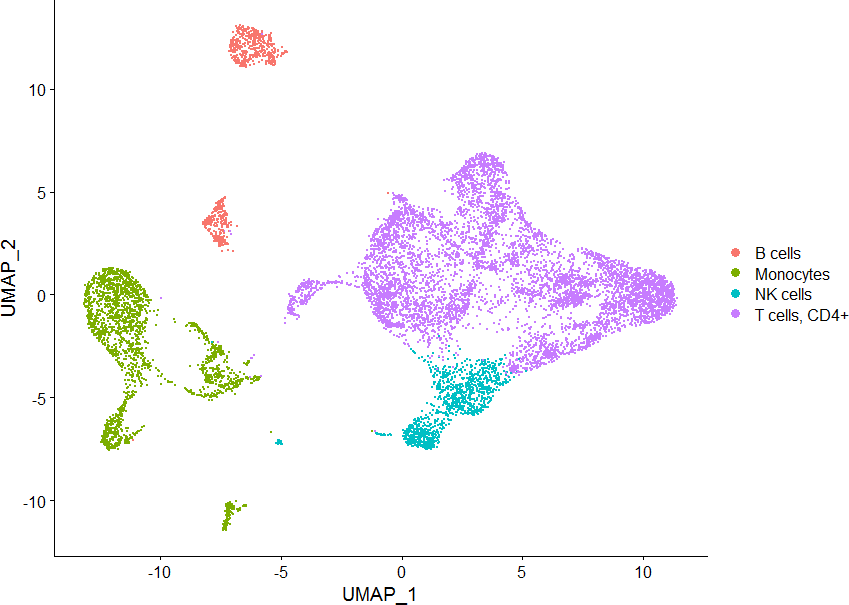
DimPlot(sce, group.by = “cellType”)
# 设置分组
sce.ob$sample[grep(“^pbmc[0-9]”, sce.ob$sample)] <- “HNC_PBMC”
sce.ob$sample[grep(“^TIL[0-9]”, sce.ob$sample)] <- “HNC_TIL”
选择配体和受体
注:ramilowski_pairs来源于软件自带的人的配体受体数据
ramilowski_pairs %>% head
ligand receptor pair
1 A2M LRP1 A2M_LRP1
2 AANAT MTNR1A AANAT_MTNR1A
3 AANAT MTNR1B AANAT_MTNR1B
4 ACE AGTR2 ACE_AGTR2
5 ACE BDKRB2 ACE_BDKRB2
6 ADAM10 AXL ADAM10_AXL
# 提取表达的配体和受体
ligs <- as.character(unique(ramilowski_pairs$ligand))
recs <- as.character(unique(ramilowski_pairs$receptor))
ligs.present <- rownames(sce.ob)[rownames(sce.ob) %in% ligs]
recs.present <- rownames(sce.ob)[rownames(sce.ob) %in% recs]
genes.to.use <- union(ligs.present,recs.present)
分组间差异分析,得到差异的配体受体
Idents(sce.ob) <- “sample”
markers <- FindAllMarkers(sce.ob, assay=”RNA”, features=genes.to.use, only.pos=TRUE)
ligs.recs.use <- unique(markers$gene)
##保留差异表达的配体-受体列表
interactions.forward1 <- ramilowski_pairs[as.character(ramilowski_pairs$ligand) %in% ligs.recs.use,]
interactions.forward2 <- ramilowski_pairs[as.character(ramilowski_pairs$receptor) %in% ligs.recs.use,]
interact.for <- rbind(interactions.forward1,interactions.forward2)
dim(interact.for)
[1] 758 3
准备celltalker的输入文件
expr.mat 基因表达数据
defined.clusters 细胞亚群信息
defined.groups样本分组信息,例如:HSC_PBMC,HSC_TIL
defined.replicates 样本信息,例如:pbmc1,pbmc2,pbmc3,TIL1,TIL2,TIL3
##准备celltalker的输入文件
expr.mat <- GetAssayData(sce.ob, slot=”counts”)
rownames(expr.mat)<-rownames(expr.mat)
defined.clusters <- sce.ob@meta.data$cellType
names(defined.clusters)<-colnames(sce.ob)
defined.groups <- sce.ob@meta.data$sample
names(defined.groups)<-colnames(sce.ob)
defined.replicates <- sce.ob@meta.data$orig.ident
names(defined.replicates)<-colnames(sce.ob)
reshape_matrices()构建 celltalker 输入数据
##重构数据,为每个样本分配相应的表达矩阵
reshaped.matrices <- reshape_matrices(count.matrix = expr.mat,
clusters = defined.clusters,
groups = defined.groups,
replicates = defined.replicates,
ligands.and.receptors = interact.for)
reshaped.matrices
# A tibble: 2 x 2
group samples
1 HNC_PBMC
2 HNC_TIL
unnest(reshaped.matrices,cols=”samples”)
# A tibble: 6 x 3
group sample expr.matrices
1 HNC_PBMC pbmc1
2 HNC_PBMC pbmc2
3 HNC_PBMC pbmc3
4 HNC_TIL TIL1
5 HNC_TIL TIL2
6 HNC_TIL TIL3
names(pull(unnest(reshaped.matrices,cols=”samples”))[[1]])
[1] “B cells” “Monocytes” “NK cells” “T cells, CD4+”
create_lig_rec_tib()为每个分组创建一致的配体-受体表达
consistent.lig.recs <- create_lig_rec_tib(exp.tib = reshaped.matrices,
clusters = defined.clusters,
groups = defined.groups,
replicates = defined.replicates,
cells.reqd = 10,
freq.pos.reqd = 0.5,
ligands.and.receptors = interact.for)
consistent.lig.recs
# A tibble: 2 x 2
group lig.rec.exp
1 HNC_PBMC
2 HNC_TIL
unnest(consistent.lig.recs[1,2], cols = “lig.rec.exp”)
# A tibble: 4 x 2
cluster.id ligands.and.receptors
1 B cells
2 Monocytes
3 NK cells
4 T cells, CD4+
putative_interactions()在给定的分组的cluster之间推断表达的ligand-receptor之间的相互作用
put.int <- putative_interactions(ligand.receptor.tibble = consistent.lig.recs,
clusters = defined.clusters,
groups = defined.groups,
freq.group.in.cluster = 0.05,
ligands.and.receptors = interact.for)
put.int
# A tibble: 2 x 2
group lig_rec_list
1 HNC_PBMC
2 HNC_TIL
识别唯一的interactions
#Identify unique ligand/receptor interactions present in each sample
unique.ints <- unique_interactions(put.int, group1 = “HNC_PBMC”, group2 = “HNC_TIL”, interact.for)
unique.ints
# A tibble: 3 x 2
comparison ligands.and.receptors
1 unique1v2
2 unique2v1
3 common
circos_plot()可视化展示
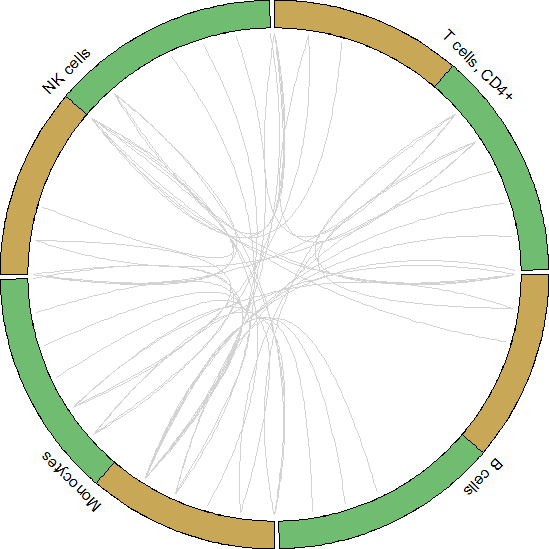
#Get data to plot circos for HNC_PBMC
pbmc.to.plot <- pull(unique.ints[1,2])[[1]]
for.circos.pbmc <- pull(put.int[1,2])[[1]][pbmc.to.plot]
circos_plot(interactions = for.circos.pbmc, clusters = defined.clusters)
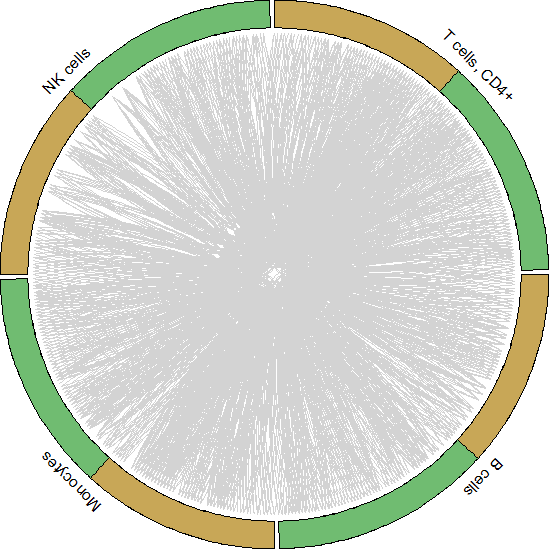
#Get data to plot circos for HNC_TIL
tonsil.to.plot <- pull(unique.ints[2,2])[[1]]
for.circos.tonsil <- pull(put.int[2,2])[[1]][tonsil.to.plot]
circos_plot(interactions=for.circos.tonsil,clusters=defined.clusters)
Celltalker搭建细胞互作通讯桥梁,为细胞互作研究提供了基础和方法,基于基因表达数据和配体-受体数据库信息可以评估细胞之间的交流。小编今天为你简答介绍单细胞数据分析之Celltalker应用,后期会为大家呈现更多的单细胞系列课程学习,欢迎大家一起学习更多关于单细胞数据处理方法和高阶分析内容。




 京公网安备 11011302003368号
京公网安备 11011302003368号