
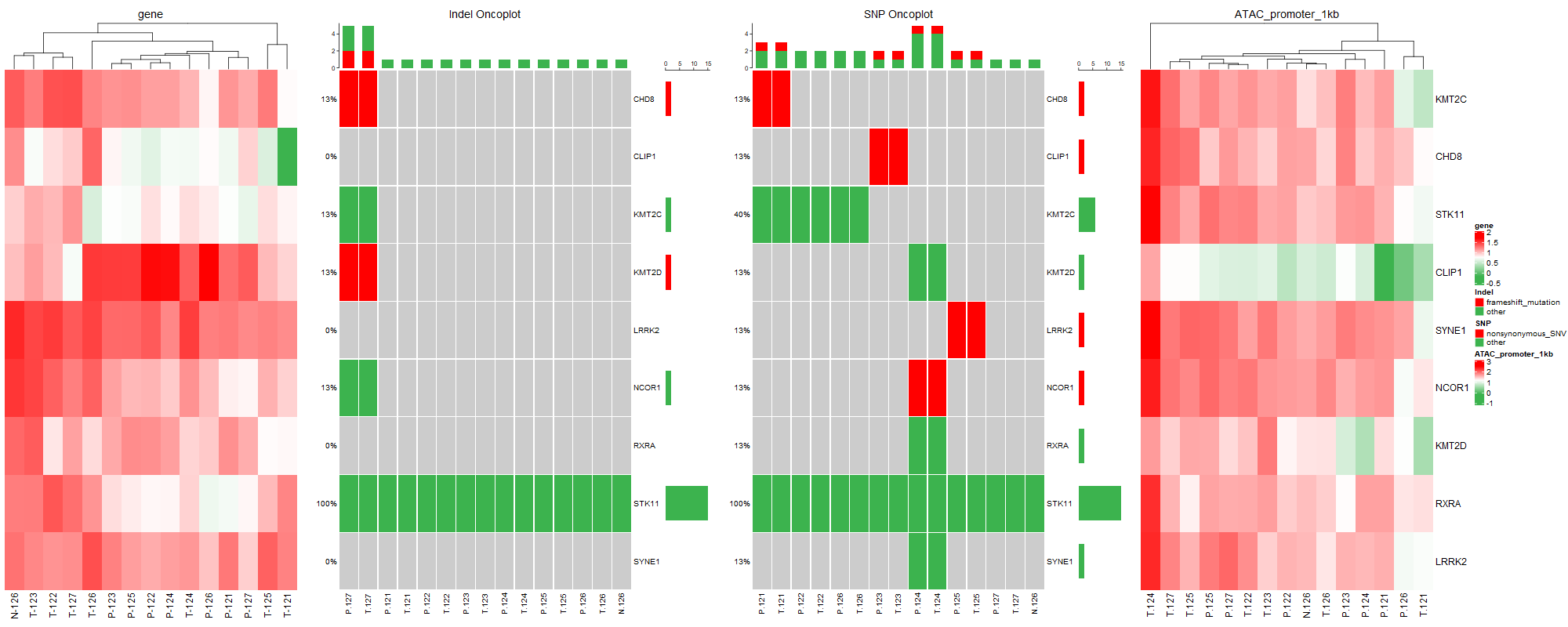
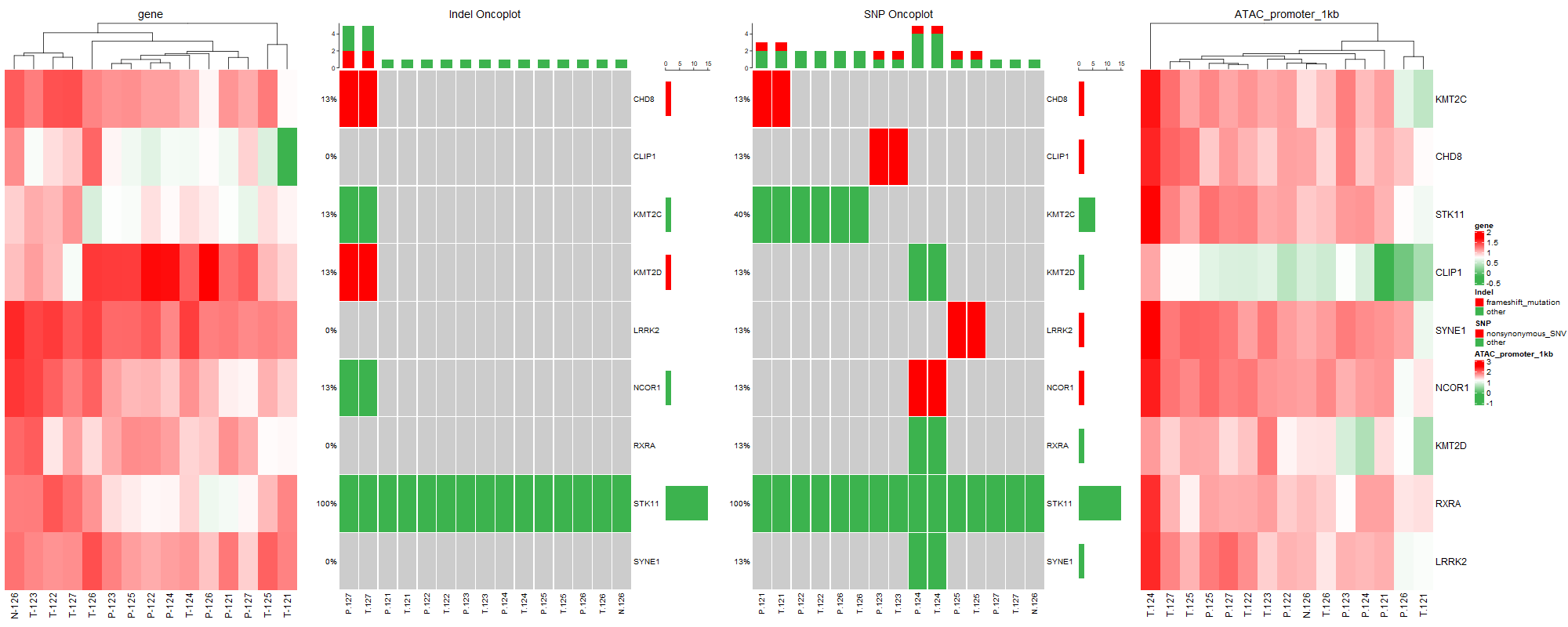
将多图组合到一起;用到R包ComplexHeatmap;
此图中包含两种基本图,一种是基因瀑布图,另外一种是热图。
脚本如下:
setwd("F:/work/个性化/基因瀑布图")
test<-read.delim("test.txt", header=TRUE ,check.names = F)
#此处读取的是四个文件的基因交集,只有一列基因,这样最后呈现的图的样本和基因个数一样。
library(ComplexHeatmap)
png("test.png",type="cairo",width=2000,height=800)
color.1 <- colorRampPalette(rev(c("red", "white", "#3CB34E")))(100)
data<- read.delim("mRNA_fpkm.list", row.names = 1, header=TRUE ,check.names = F)
row.names(data)<-data$Symbol
data<-data[which(rownames(data)%in%test$gene_symbol),]
data= data[, -1]
data <- log10(data+0.000001)
data<-as.matrix(data)
ht1<-Heatmap(data,col=color.1,cluster_columns = T,cluster_rows = F,name="gene",column_title = "gene",show_row_names = T,show_column_names = T)
#此处既显示样品名又显示基因名
col<-c("frameshift_mutation"="red","other"="#3CB34E","nonsynonymous_SNV"="red")
alter_fun<-list(
background=function(x,y,w,h){
grid.rect(x,y,w-unit(0.5,"mm"),h-unit(0.5,"mm"),
gp=gpar(fill="#cccccc",col=NA))
},
frameshift_mutation=function(x,y,w,h){
grid.rect(x,y,w-unit(0.5,"mm"),h-unit(0.5,"mm"),
gp=gpar(fill=col["frameshift_mutation"],col=NA))
},
other=function(x,y,w,h){
grid.rect(x,y,w-unit(0.5,"mm"),h-unit(0.5,"mm"),
gp=gpar(fill=col["other"],col=NA))
},
nonsynonymous_SNV=function(x,y,w,h){
grid.rect(x,y,w-unit(0.5,"mm"),h-unit(0.5,"mm"),
gp=gpar(fill=col["nonsynonymous_SNV"],col=NA))
}
)
library(do)
mat <-read.table("indel_keygene.xls",head=T,sep="\t",stringsAsFactors = FALSE)
#此处导入处理后的indel文件,格式如图:
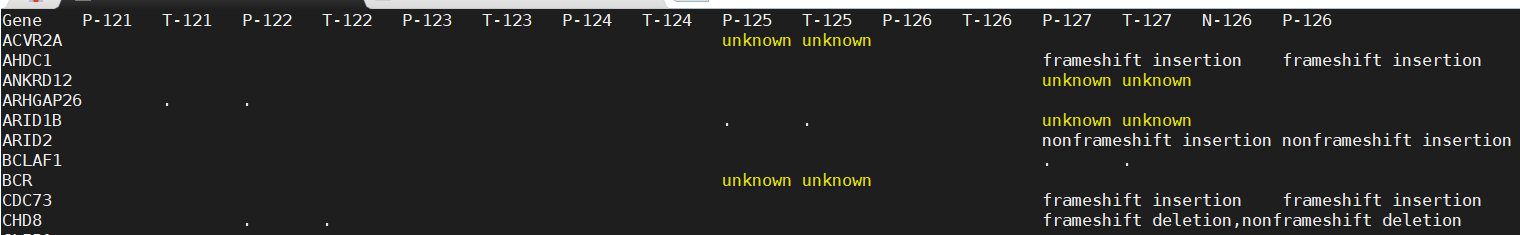
rownames(mat)<-mat$Gene
mat<-mat[which(rownames(mat)%in%test$gene_symbol),]
mat<-mat[,-1]
mat[is.na(mat)]<-""
mat<-Replace(mat,from = "^.$",to = " ",pattern = "")
#此处Replace是R包do中的函数,将文件中的内容替换成自己所需要的内容
mat<-Replace(mat,from = "^,.",to = " ",pattern = "")
mat<-Replace(mat,from = ",.$",to = " ",pattern = "")
mat<-Replace(mat,from = "^.,",to = " ",pattern = "")
mat<-Replace(mat,from = "nonframeshift deletion,frameshift deletion",to = "frameshift_mutation",pattern = "")
mat<-Replace(mat,from = "frameshift deletion,nonframeshift deletion",to = "frameshift_mutation",pattern = "")
mat<-Replace(mat,from = "nonframeshift deletion",to = "other",pattern = "")
mat<-Replace(mat,from = "nonframeshift insertion",to = "other",pattern = "")
mat<-Replace(mat,from = "frameshift deletion",to = "frameshift_mutation",pattern = "")
mat<-Replace(mat,from = "frameshift insertion",to = "frameshift_mutation",pattern = "")
mat<-Replace(mat,from = "unknown",to = "other",pattern = "")
#oncoPrint(mat)
#指定变异类型的标签,和数据中的类型对应
heatmap_legend_param<-list(title="Indel",at=c("frameshift_mutation", "other"),labels=c("frameshift_mutation", "other"))
#设定标题
column_title<-"Indel Oncoplot"
ht2<-oncoPrint(mat,alter_fun = alter_fun,col=col,column_title=column_title,heatmap_legend_param = heatmap_legend_param,remove_empty_columns = F,remove_empty_rows = F,show_column_names = T,show_row_names =T)
mat <-read.table("keygene.SNP.xls",head=T,sep="\t",stringsAsFactors = FALSE)
#导入SNP文件,格式如图
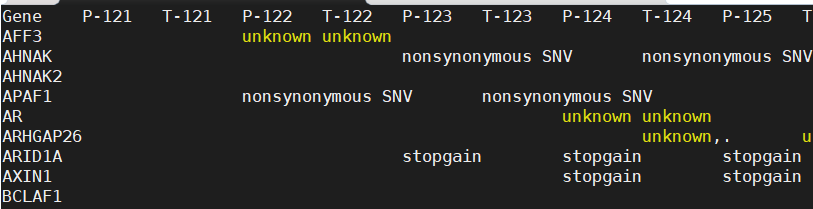
rownames(mat)<-mat$Gene
mat<-mat[which(rownames(mat)%in%test$gene_symbol),]
mat<-mat[,-1]
mat[is.na(mat)]<-""
#mat<-str_replace("nonframeshift insertion","other","")
mat<-Replace(mat,from = "^.$",to = "",pattern = "")
mat<-Replace(mat,from = "^,.",to = "",pattern = "")
mat<-Replace(mat,from = ",.$",to = "",pattern = "")
mat<-Replace(mat,from = "^.,",to = "",pattern = "")
mat<-Replace(mat,from = "stopgain",to = "other",pattern = "")
mat<-Replace(mat,from = "stoploss",to = "other",pattern = "")
mat<-Replace(mat,from = "unknown",to = "other",pattern = "")
mat<-Replace(mat,from = "^synonymous SNV$",to = "other",pattern = "")
mat<-Replace(mat,from = "^nonsynonymous SNV$",to = "nonsynonymous_SNV",pattern = "")
mat<-Replace(mat,from = "synonymous SNV,nonsynonymous SNV",to = "nonsynonymous_SNV",pattern = "")
mat<-Replace(mat,from = "nonsynonymous SNV,synonymous SNV",to = "nonsynonymous_SNV",pattern = "")
#oncoPrint(mat)
mat<-as.matrix(mat)
#指定变异类型的标签,和数据中的类型对应
heatmap_legend_param<-list(title="SNP",at=c("nonsynonymous_SNV", "other"),labels=c("nonsynonymous_SNV", "other"))
#设定标题
column_title<-"SNP Oncoplot"
ht3<-oncoPrint(mat,alter_fun = alter_fun,col=col,column_title=column_title,heatmap_legend_param = heatmap_legend_param,remove_empty_columns = F,remove_empty_rows = F,show_column_names = T,show_row_names =T)
data<- read.delim("ATAC_promoter_1kb.xls", row.names = 1, header=TRUE ,check.names = F)
row.names(data)<-data$gene_symbol
data<-data[which(rownames(data)%in%test$gene_symbol),]
data= data[, 5:19]
data[is.na(data)]<-0
data<-data[which(rowSums(data)>0),]
data <- log10(data+0.000001)
data<-as.matrix(data)
library(ComplexHeatmap)
color.1 <- colorRampPalette(rev(c("red", "white", "#3CB34E")))(100)
ht4<-Heatmap(data,col=color.1,cluster_columns = T,cluster_rows = F,name = "ATAC_promoter_1kb",column_title = "ATAC_promoter_1kb",show_row_names = T,show_column_names = T)
draw(ht1+ht2+ht3+ht4,gap=unit(0.8,"cm"))
dev.off()
其中需要注意的是当有多个基因瀑布图时,alter_fun和col需在同一条命令中设置。通过以上一顿操作,快速将基因瀑布图和热图组合在一起,既展示了基因的结构变异、开放状态、基因表达,呈现多组学的角度探讨疾病发生发展。这不就完成了Fig1还升华了Fig1的深度。
再多的操作文档,总是不如实际操作来得理解深刻。百迈客云平台(BMKCloud)实现你零基础分析的所有梦想!52款APP实现高度自由个性化分析,118款工具、交互的个性化分析提供无上限分析实操,云平台课堂高效助您方案设计和信息分析(http://www.biocloud.net/),百迈客云成为您的私人信息分析平台,让基因分析更简单。
所以大家快快动起手来,在实际操作中理解学习,若在学习中有不懂或疑问的地方,您可以点击下方按钮与我们联系,我们可以提供相应的分析服务。





 京公网安备 11011302003368号
京公网安备 11011302003368号