
分类: 医学研究


导读
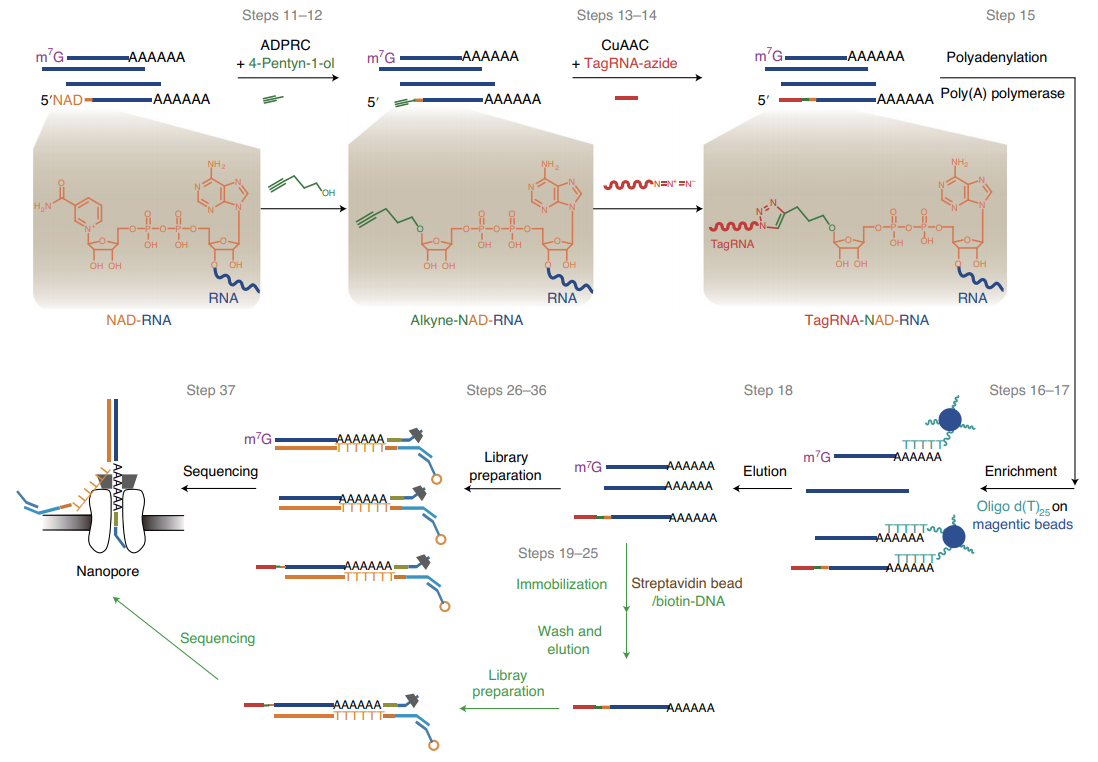
图1 NAD tagSeq的流程
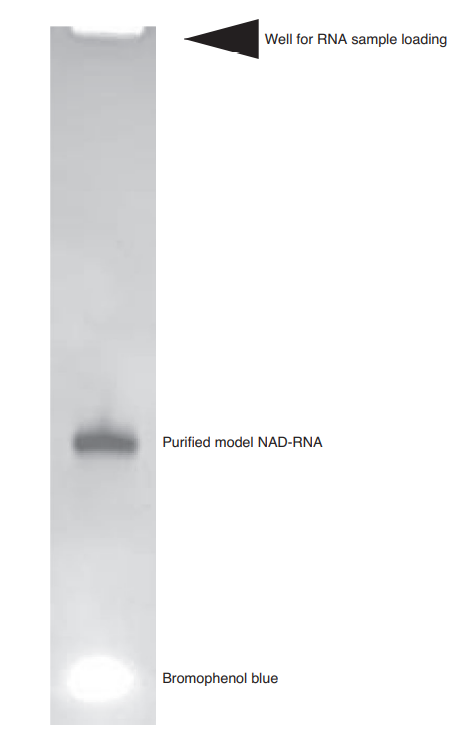
图2 纯化的model NAD-RNA的凝胶电泳
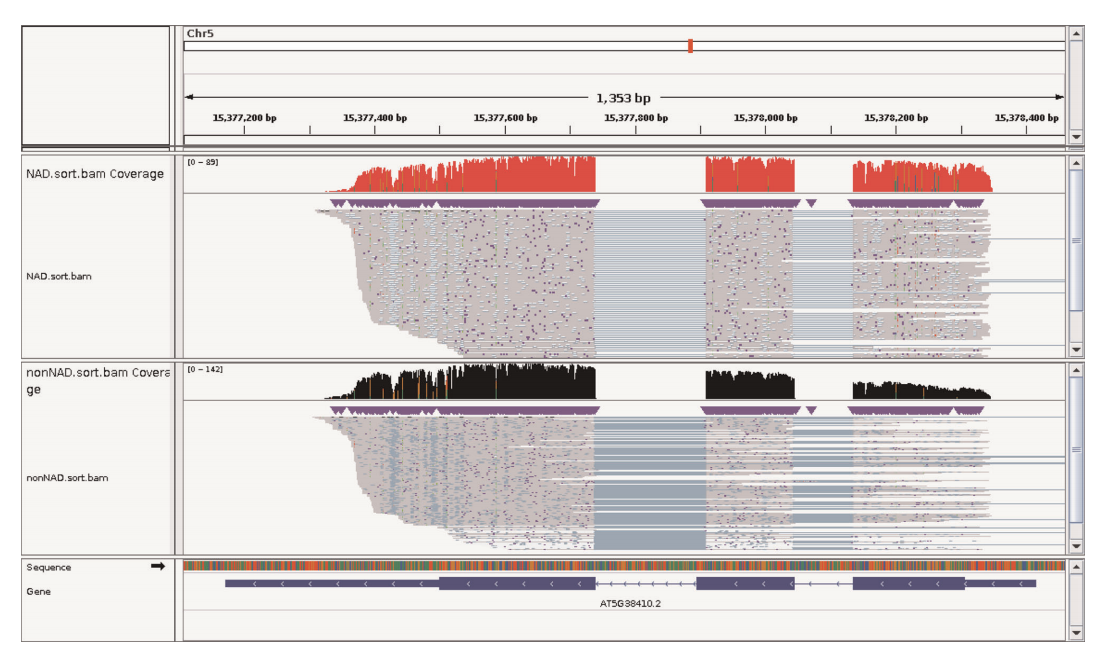
图3 NAD-RNA和noncapped-RNA reads可视化图
第四,NAD tagSeq可以揭示NAD-RNA的整体序列。ONT测序技术允许将RNA分子从3’端到5’端测序。因此,可以揭示RNA分子的序列。它允许分析cap如何通过转录后修饰(如选择性剪接或5’非翻译区)调节RNA功能。
第五,将来可能会应用通过合成RNA tag标记NAD-RNA然后进行直接RNA测序的想法,以开发类似的方法来鉴定其他NICN-capped RNA,可以开发特定的酶和/或点击化学反应来标记其他NCIN capped RNA。
局限性:纳米孔测序从3’端到5’端,有时会导致reads被截断,这意味着RNA的5’区域可能未测序。因此,某些没有RNA tag的reads可能来自NAD-RNA,这会引入一些假阴性。
此外,如果在标记之前不进行聚腺苷酸化步骤,RNA的断裂(例如在铜离子存在下的点击化学反应期间)可能会导致假阴性。
NAD tagSeq的另一个缺点是,由于ONT芯片的成本很高,它比NAD captureSeq更为昂贵。此外,Oxford Nanopore测序平台在对小RNA进行测序方面存在局限性。获得的短测序reads(不包括poly(A)尾部和RNA标签序列)约为80个核苷酸,这增加了一些小NAD-RNA可能被该分析遗漏的可能性。推测Illumina测序也可用于NAD tagSeq。但是,还需要进一步测试逆转录酶是否可以通过RNA tag和NAD cap之间的joint以到达RNA tag区域。
总结
最近文章




 京公网安备 11011302003368号
京公网安备 11011302003368号