
导读
研究背景:
人乳头瘤病毒(HPV)是宫颈癌的主要原因,HPV16和18是全球两种流行的高危HPV类型。宫颈癌是女性中第二大常见的恶性肿瘤。每年有570,000名女性被诊断出患有宫颈癌,并且有311,000名女性死于这种疾病。尽管已知持续的HPV感染会导致宫颈癌,但目前尚不清楚HPV如何诱发癌变,以及在该过程中到底发挥什么重要的功能。HPV感染后,HPV蛋白E6和E7被表达,其抑制肿瘤蛋白p53和视网膜母细胞瘤蛋白,破坏细胞增殖和许多其他生物学过程,并诱发宫颈癌。此外,HPV DNA也可能整合到人类DNA中,这是致癌过程中的早期重要事件。另外,癌症的进展可以通过病毒DNA整合入抑癌基因来解释。这种整合入宿主DNA会使那些基因失活,从而导致生长不受控制。了解病毒的致癌作用对于HPV阳性癌症的临床治疗至关重要。
结论:
作者通过Nanopore和Illumina测序对先前发表的样品的整合位点进行了重新测序。在对结果进行分析之后,得出了三点结论:首先,从所有三个数据集(即,Nanopore,Illumina和已发布的数据)中找到了19个先前发布的整合位点中的13个,表明这些元素之间具有很高的重叠率和可比性;其次,与先前发表的论文相比,Nanopore和Illumina数据确定了66个独特的整合位点,其中13个已通过Sanger测序验证,这表明与公开数据相比,作者的结果对整合位点的检测灵敏度更高;第三,作者建立了可用于通过纳米孔测序数据检测HPV整合位点的pipeline,并且无需进行纠错分析。总而言之,与现有的Illumina数据分析pipeline方法相比,测试了一种新的纳米孔数据分析方法,并证明了该方法在整合位点检测中是可靠的,所需的测序数据更少。它提供了有力的证据和工具来支持纳米孔在病毒状态识别中的潜在应用。
材料与方法
实验结果
一、数据质控
作者通过对目标区域捕获后进行文库构建,分别进行Nanopore和Illumina测序,分别获得130.2 Mb和1.64 Gb的clean reads(图1)。
图1
二、通过Nanopore和Illumina数据鉴定HPV整合位点
作者构建Nanopore分析pipeline(图2A),通过Blast与人和HPV16病毒基因组进行比对,共识别339个整合位点,通过过滤筛选最终剩余60个位点进行后续的分析。Illumina测序共获得1718个整合位点,筛选reads覆盖度大于3的位点54个进行后续分析。
图2
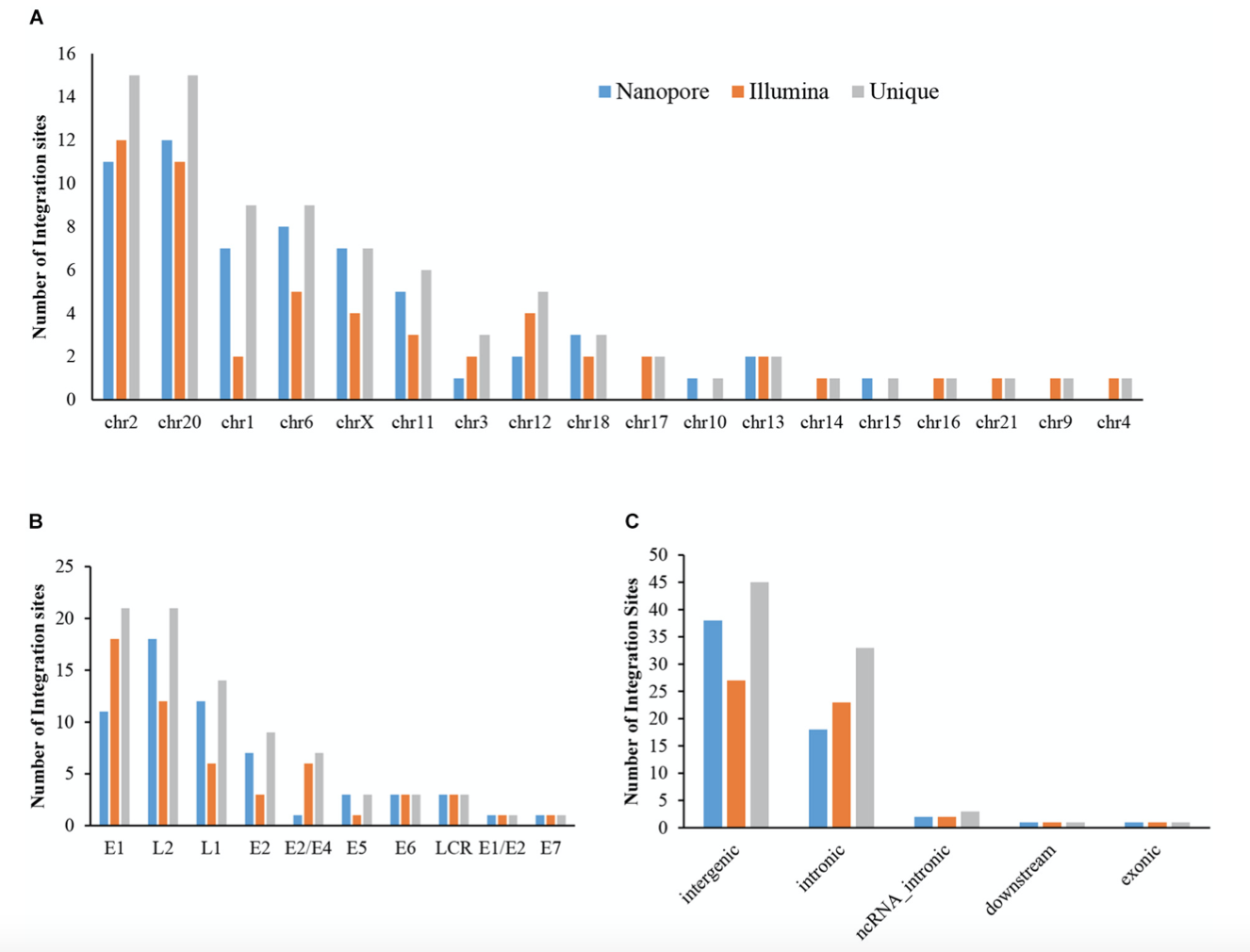
所有断点在染色体上的分布见图3A。染色体chr20、chr2、chr6分别由12、11和8个断点,chr1和chrX均存在7个断点,其余染色体断点分布均在5以下。在每个位点上的reads覆盖度从2~406不等,大约有31.7%以上的位点覆盖深度在10以上。在top5的覆盖深度位点中3个位于chr20(HPV16:2804,chr20:32516985 (406 reads);HPV16:7139,chr20:32478733(169 reads);HPV16:4276,chr20:32502143(51reads)),2个位于chrX(HPV16:5534,chrX:20464412 (132 reads);HPV16:3163,chrX: 20462930 (50 reads))。
图3B展示了60个位点在HPV基因组上的分布,L2基因中有18个,L1中有12个,E1中有11个,E2中有7个,其他每个基因(E6,LCR,E5和E7)上小于4个。
在人基因组上,有38个位点在基因间区,18个位于内含子区域,2个位于非编码区域,1个位于外显子区域,1个位于downstream区域。
进一步分析人基因组上的位点分布,在chr20染色体上的12个位点中,有10个在基因CHMP4B和RALY-AS1之间,有一个位于KIF3B和ASXL1之间,最后一个位于基因ATRN的内含子区域。除了chr20染色体外,其他染色体上也有明显的位点聚集趋势,例如,chrX染色体上的6个位点在基因RPS6KA3和CNKSR2之间,chr6染色体上的3个位点位于基因CAGE1的内含子或者downstream区域。
图3
三、在不同数据集中的位点差异
在Illumina测序获得的54个整合位点中,有19个与之前报道的相符。整合所有位点并做韦恩图(图4),有13个位点是3种数据(Nanopore、Illumina、published)都存在的,这表明不同平台间重复性很好。共有18个位点同时出现在Nanopore和Illumina中,三个位点同时出现在纳米孔测序和以前发表的论文中,只有一个位点在Illumina和已发表的论文重叠。在两个平台中确定的整合位点中,一些断点具有高度丰富的reads数,例如HPV16:2804,chr20:32516985,Nanopore结果中有406个reads,Illumina结果中有1439个reads。其他断点的reads数很少,例如HPV16:4250,chr21:97550764,其中Nanopore结果中有两个reads,而Illumina结果中只有四个reads。
除了重叠的位点外,每个平台都有自己独特的整合位点。 Nanopore、Illumina和文献中,分别拥有26、22和2个唯一的整合位点。在Nanopore识别的26个位点中,其中6个reads支持度在6以上。因此,在Illumina,Nanopore和文献这三种方法确定的整合位点中,Nanopore具有相对可靠的丰度,可以确定准确的整合位点。
图4
作者为了测试数据确定的新整合位点的准确性,利用Sanger测序进行验证。总共选择了14个整合位点进行验证,并对13个进行PCR扩增并成功测序,表明这些整合位点的真正阳性,这些阳性位点大多由Nanopore和Illumina平台鉴定。
图5
四、受影响的基因功能分析
合并3种不同平台的整合位点,并对整合完的83个位点进行注释,并进行了进一步的功能分类和途径分析。这些基因分为八个生物学过程,包括RNA聚合酶II启动子的转录正调控(6个基因),RNA聚合酶II启动子的转录负调控(6个基因),RNA聚合酶II启动子的转录调控(5个基因), RNA聚合酶II启动子的转录(4个基因),视黄酸受体信号传导途径的负调控(2个基因),皮肤屏障的建立(2个基因),近端/远端模式形成(2个基因)和胚胎肢体形态发生(2个基因)六个胞质(13个基因),核质(8个基因),胞外外泌体(8个基因),蛋白质结合(6个基因),DNA结合(2个基因)和肌动蛋白结合(2个基因)。
讨论
数据表明,大多数整合位点存在于人类基因组的基因间区域中,与先前的研究一致。由于鉴定出人类基因组中有很大一部分(60%)的基因间区域,研究结果确定了约40%的整合位点位于人类基因组的基因间区域中,没有显着差异。因此,可以得出结论,整合位点在人类基因组中的分布方式受到基因组功能结构性质的影响。另外作者还发现整合发生在非编码RNA中,它起着许多重要的功能,例如lncRNA与p53蛋白相互作用。插入ncRNA可能会导致更严重的功能中断。其中HPV有插入的倾向,例如肿瘤蛋白63(TP63),它在致癌作用中起着非常重要的作用。
在这项研究中,作者确定了两个基因CHMP4B和RALY-AS1。整合簇的趋势非常强,在76个独特的整合位点以及其他几个染色体区域中有10个整合位点。为什么病毒会整合到几个特定区域,并且是随机的还是特定的?需要进一步的研究来回答这些问题。




 京公网安备 11011302003368号
京公网安备 11011302003368号