

英文标题:Culture-enriched metagenomic sequencing enables in-depth profiling of the cystic fibrosis lung microbiota
机构:加拿大卡尔加里大学微生物学、免疫学和传染病学系
发表期刊:Nature Microbiology
影响因子:15.54
发表时间:2020 Jan 20
摘要
作者将扩增子测序与富集培养宏基因组测序相结合研究人类微生物群落。以肺囊性纤维化为例,平均培养了82.13%的可操作分类单元(OTUs),占痰液样本直接测序所鉴定的相对丰度的99.3%,更重要的是,与直接测序相比,富集培养方法鉴定的OTUs多了63.3%。作者开发了平板覆盖算法(PLCA),以确定富集培养平板的代表性子集,从而在该子集上进行宏基因组研究。与不依赖培养的方法相比,可以恢复更多的物种多样性(包含更好的低丰度物种的组装基因组,MAGs),更长的重叠群和更好的功能注释结果。PLCA算法也可以应用于此前发布的肠道菌群数据集,富集培养分子图谱可以更好的了解微生物菌群在人类健康和疾病中的作用。
背景介绍
扩增子测序(如16S rRNA基因)简单快速地鉴定出微生物菌群组成和相对丰度,而宏基因组测序除了物种之外,还可以评估群落的基因组分、潜在功能,测序数据的有效性,取决于短序列拼接成重叠群的情况。拼接质量受菌群的复杂度、测序技术、宿主DNA污染比例等影响。宏基因组测序对于宿主DNA比例较高的样本类型(如皮肤、组织、呼吸道)来讲是一个挑战。
实验方法
基于作者之前的研究(肺囊性纤维化患者的痰液样本富集培养扩增子测序鉴定到48科细菌,其中43科可以培养),收集到的样本分别在有氧和厌氧条件下,经过13种不同的培养基富集。同时,对这些样本直接进行16S rRNA测序,富集培养后的共26个样本也进行16S测序,同时挑选了8个富集后的样本进行宏基因组测序。
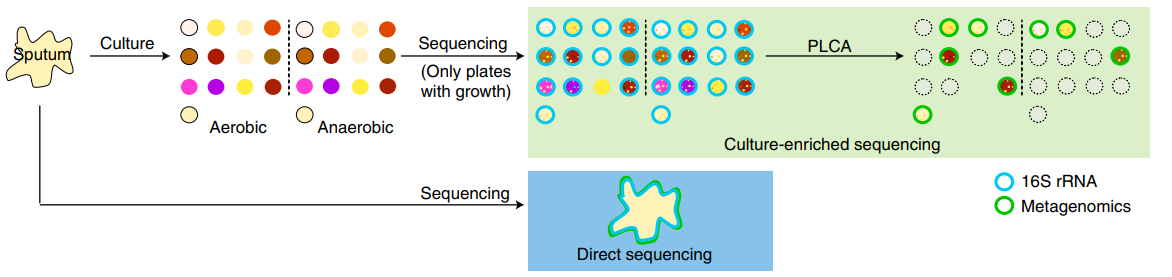
图1 富集培养宏基因组测序工作流程
实验结果
肺囊性纤维化痰液微生物菌群大多是可培养的,富集培养增加OTU恢复率
作者判断OTU可培养的条件:1)至少10条序列;2)至少1个富集板中回收到且丰度比例至少为0.01%。直接测序鉴定到的OTU中82.13%是可培养的,这些可培养的在所有直接测序的样本中平均丰度比例为99.3%(图2a),富集培养扩增子测序比直接测序获得更多的OTUs(图2b),如样本1恢复到49个OTUs,其中42个在富集培养中也恢复到,富集培养还恢复了额外的124个OTUs(图2a)。
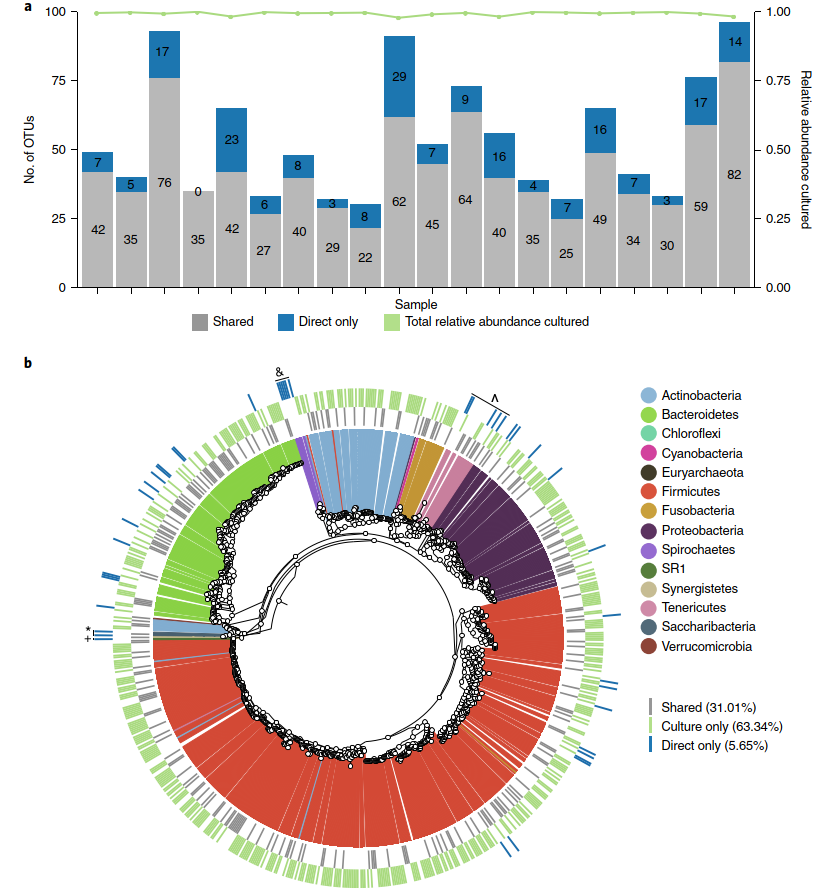
图2 肺囊性纤维化痰液微生物菌群大多是可培养的
培养OTU恢复率取决于培养基类型和是否有氧
培养基类型和培养环境对于微生物菌群多样性是重要的。富集培养和直接16S测序结果中物种分布和β多样性结果表明有氧和厌氧条件促进了不同的物种恢复(图3a,b)。不同培养基类型的物种进行聚类显示选择性培养和非选择性培养的重要性(图3c),有些属的生长模式也存在OTU依赖差异(如普氏菌属,图3d)。作者在临床实验室常用的4种典型培养条件的基础上,扩展了培养条件,获得将近2倍的OTUs(图3e)。富集培养的另一个优点是可以从冻存细菌中回收感兴趣的微生物。嗜麦芽窄食单胞菌从两个冻存的脱脂牛奶块中分离得到,其相对丰度比例很低(1.3%和1.5%),牛奶块在特定的培养基类型培养(图3f)。
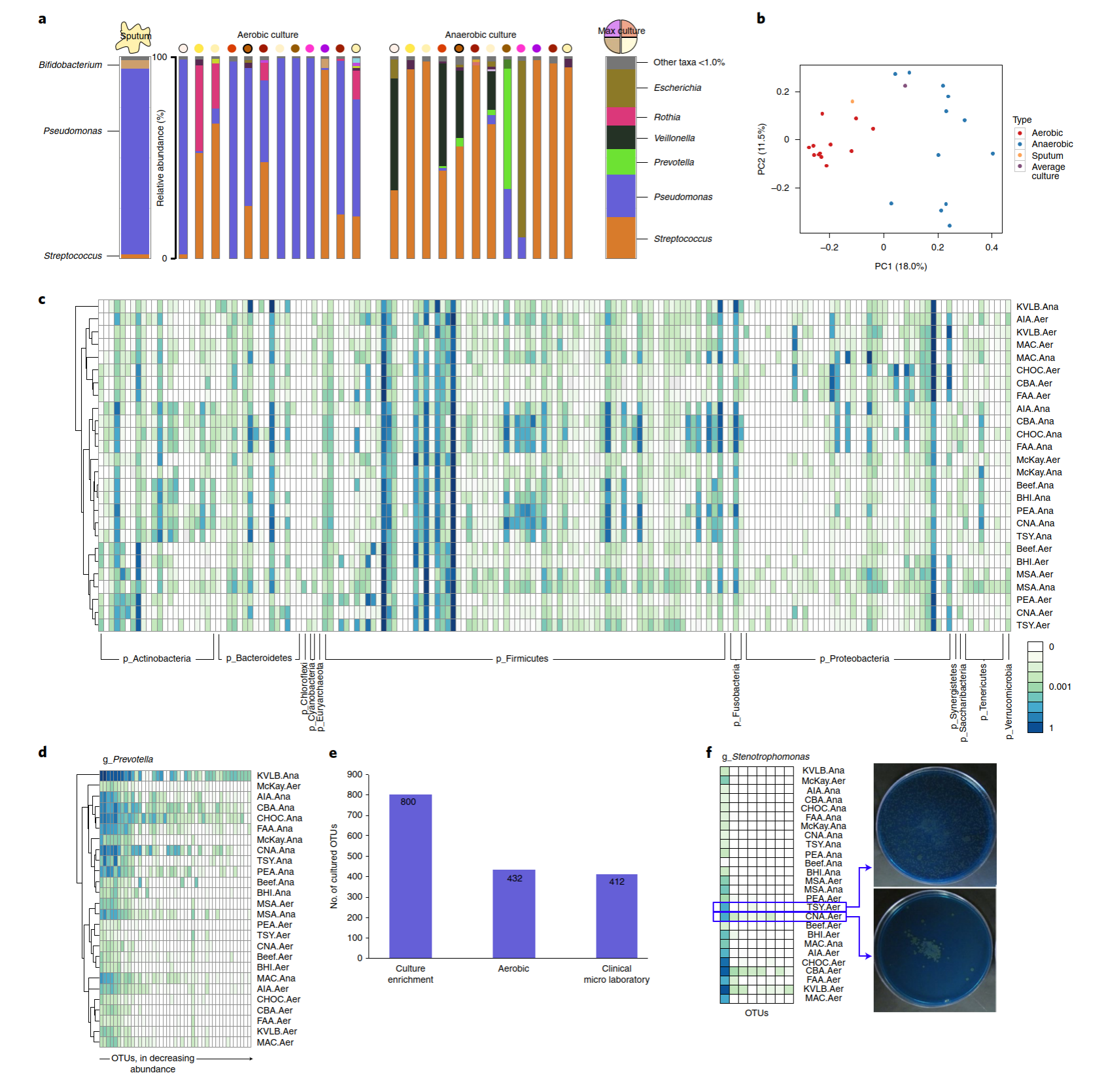
图3 通过培养富集物种分类多样性增加
PLCA算法为培养富集的宏基因组测序提供信息
肺部蕴藏了微生物菌群,每个个体是有差异的,虽然不同的培养条件是捕获每个个体多样性的必要条件,但是并不意味着收集到的每个样品都需要进行培养富集,这对其他人体部位菌群同样适用,如肠道中有很多微生物在人群中普遍存在,而有些微生物则对某些个体来讲是特异的。我们不能预先知道哪些样品可以体现菌群状态,但我们可以采用16S rRNA测序,以确定菌群物种分布,来选择哪些样品适合进行富集培养。
作者采用了PLCA算法,来决定需要进行富集培养宏基因组测序的最小样品数,算法对任何含有大部分可培养富集的菌群生态均适用。PLCA算法有两个版本,从头 PLCA不依赖于直接测序结果再现富集培养菌群,而校正PLCA侧重于直接测序结果中OTU的修复,这取决于是否都对原始样品的菌群组成感兴趣。采用这两种算法,数据集凸显了不同样本的独特性:从头PLCA显示,每个样本具有独特的平板集,并且每个培养条件对每个样本都是必要的;校正PLCA显示对数据集中样本进行富集培养宏基因组测序是很有必要的(图4a,b)。
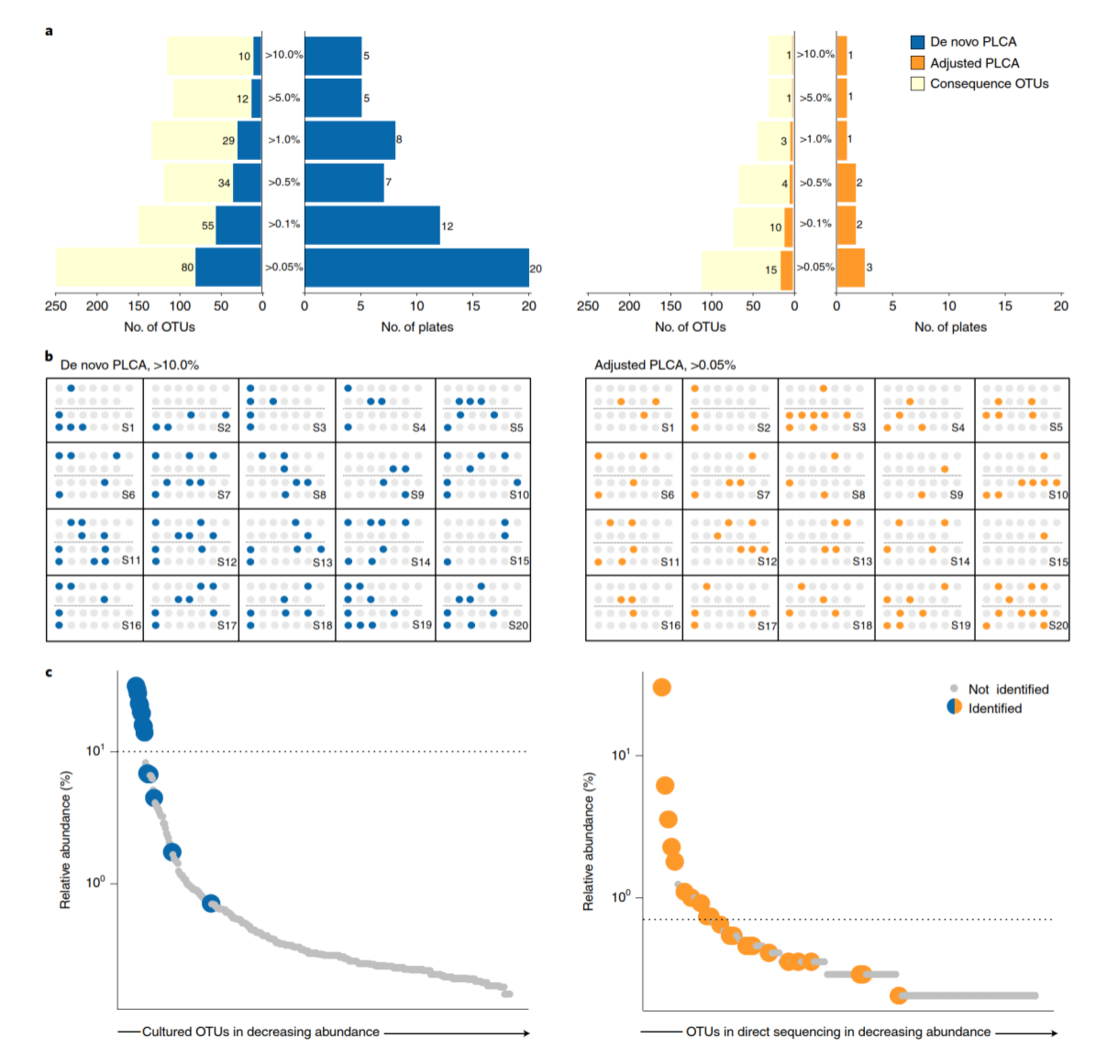
图4 PLCA算法确定了富集培养宏基因组测序的最佳平板组
富集培养宏基因组测序提供了更好的分类和功能分辨率
作者选择代表性样品,同时采用PLCA算法的两个版本。从头PLCA和校正PLCA分别显示需要进行5个和3个富集培养平板的宏基因组测序。通过宏基因组样本合并组装、binning和物种注释,比较两种算法的实验结果和预期结果。从头PLCA恢复10个bins,与PLCA阈值上的10个OTUs物种比对结果一致,另有6个阈值以下的物种,校正PLCA恢复了预期10个预期的OTUs,另有14个阈值下的OTUs(图4c)。
对从头PLCA、校正PLCA和直接宏基因组测序的数据集分别进行合并组装、binning,定义完整度≥70%和污染度<10%的为MAG,阈值下为non-MAG。其中从头校正PLCA得到7MAGs和9个non-MAG bins,校正PLCA得到12个MAGs和12个non-MAG bins,而直接宏基因组测序仅得到1个MAG和1个non-MAG bin(图5)。直接测序的序列比对富集培养的bins,在从头PLCA和校正PLCA中均只在1个bin中观察到显著覆盖(图5,蓝点)。直接测序获得2个bins均注释到假单胞菌的种,富集培养的bins注释物种多样性增加(14个属,图5)
通过富集培养获得的分类学多样性的增加直接转化为该微生物群落功能信息的增加(图6)。富集培养测序在直系同源蛋白簇(COGs)、毒力基因、抗生素抗性基因、CRISPRs和次级代谢功能方面获得更大的多样性和功能鉴定数量。先前的研究已经证实囊性纤维化气道以及整个人类微生物群中存在单一物种的异质性群体。作者通过鉴定基因单倍型来计算每个宏基因组bin的遗传变异性(图6d)。在一些宏基因组bins中,作者鉴定到一个一致的和少量的基因单倍型,表示这个bin代表一个单一的基因组群体(即一个菌株)。在大多数bins中,作者观察到大量的单倍型多样性,指示异质性群体,即多菌株。不出所料,这些区域中每个基因的单倍型数量是多种多样的,这表明细菌基因组内进化压力的已知范围。平均而言,假单胞菌属种中鉴定出更多的基因单倍型,富集培养的bins比直接测序更多。然而,直接测序鉴定了大量的普遍存在的单倍型基因离群值。Bin的完整性与平均单倍型频率没有相关性。
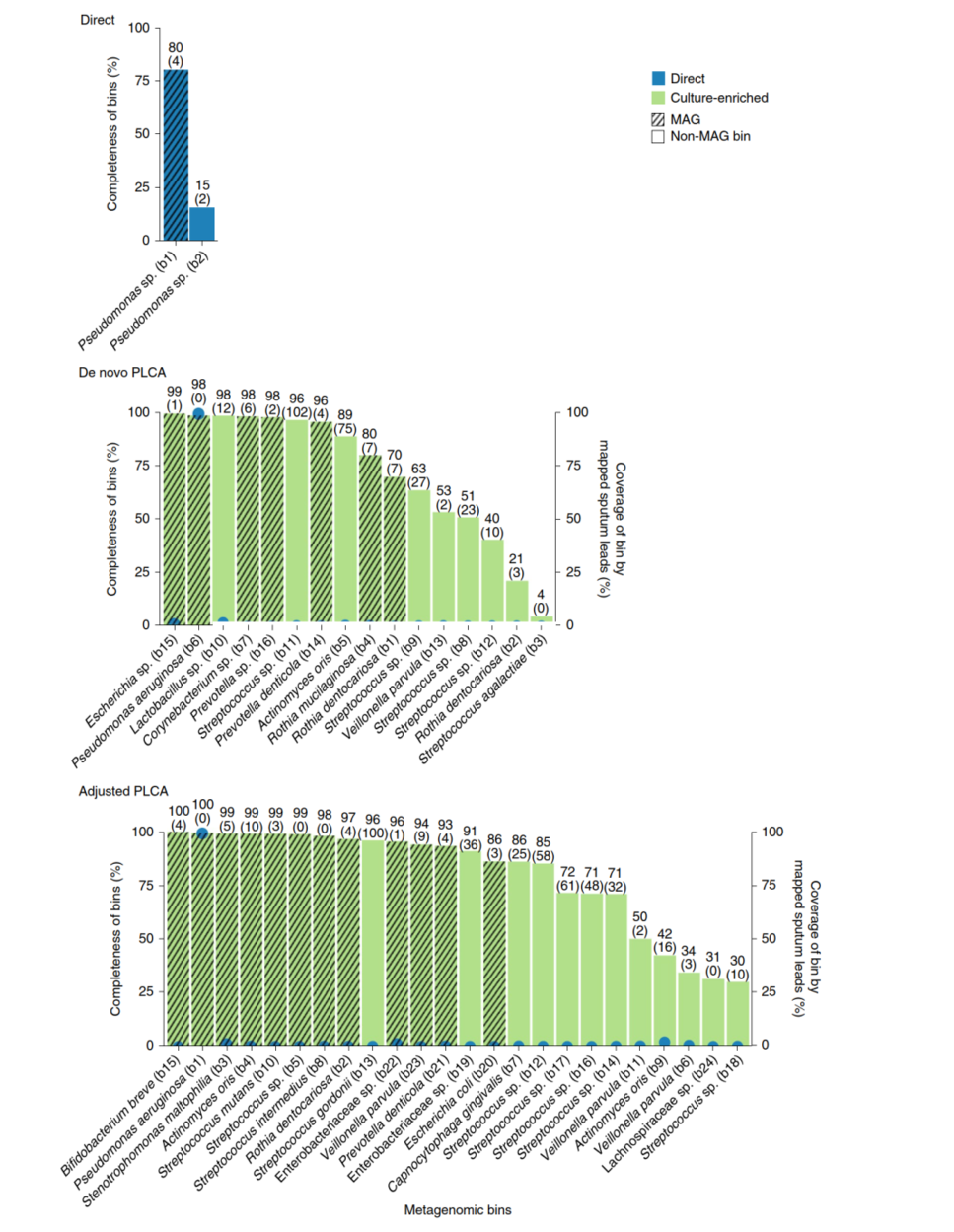
图5 项目数据中第一批样本直接测序和腹肌培养测序的MAG和non-MAG bins结果
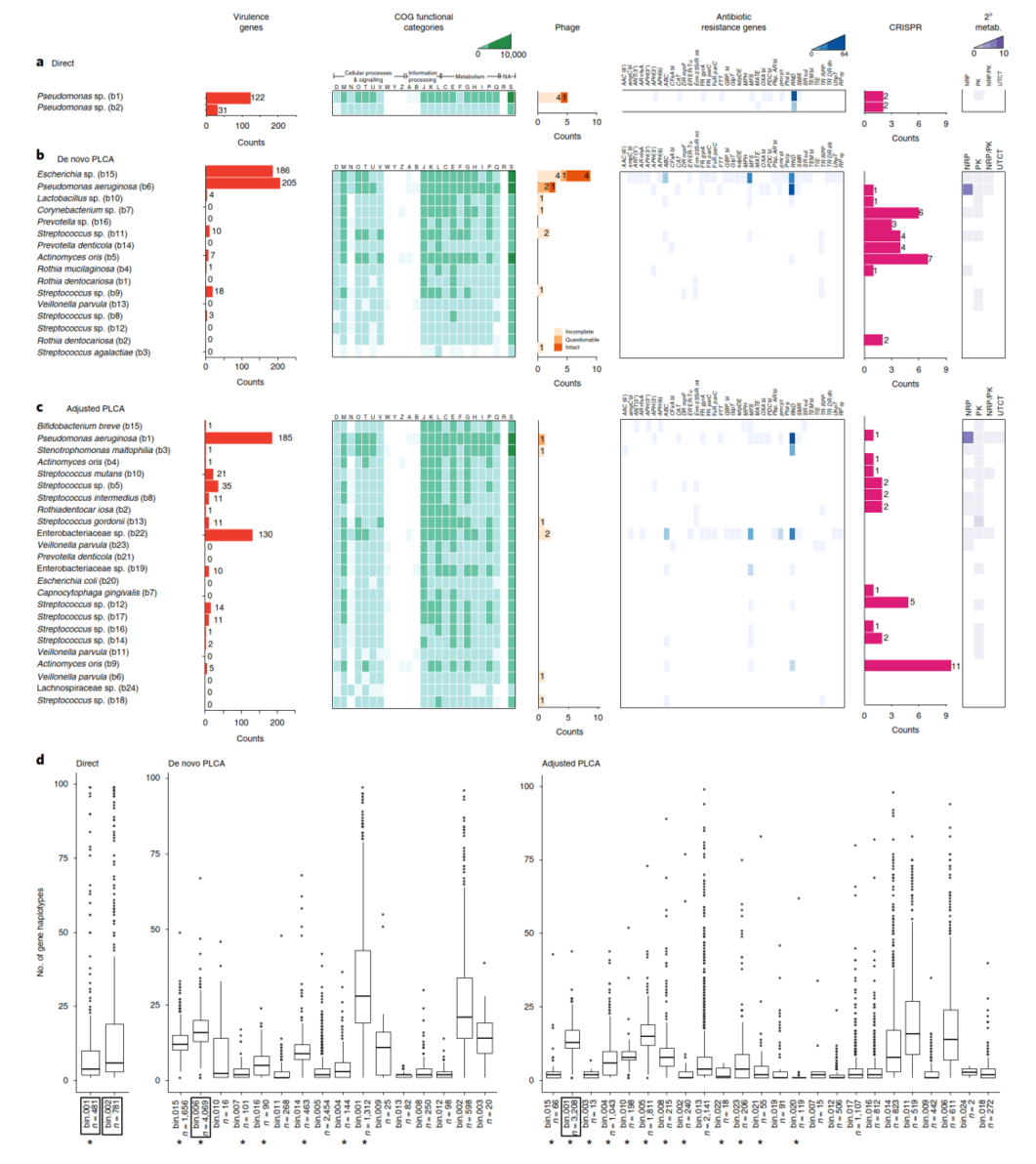
图6 直接和富集培养宏基因组测序的物种和功能多样性结果
讨论
测序成本的降低和测序技术的增加,彻底改变了我们研究人类微生物群的方式,从而使人们对群落及其与健康和疾病之间的关系有了更深入的了解。肠道是目前研究最深入的人体微生物组,很多研究将其与各种疾病和状况联系在一起。皮肤、活检组织、口腔微生物样本等许多重要的与人类相关的群落具有高比例的非微生物DNA,这些样本的宏基因组测序数据由于宿主污染,必须通过比对过滤掉大部分序列,意味着只有群落中高丰度比例的物种可以被组装成MAGs。作者论证了与传统培养技术相关联的富集宏基因组测序可以提高群落的分辨率,并且为了最有效的结合富集培养和直接测序,作者设计了PLCA算法,其确定了对于重现原始菌群(扩增子测序),富集培养宏基因组测序很有必要。
富集培养测序与直接测序相结合观察到更多的物种多样性并且更深入的了解与人类相关的微生物群落,更好的了解每种微生物的基因组成。此外,将这些微生物进行培养,我们可以进行体外机制的研究,并且可以在人类健康和疾病的背景下,更好的了解这些群落。



 京公网安备 11011302003368号
京公网安备 11011302003368号