
众所周知,要获得基因组的完整图片,就必须组装reads,以目前主要的测序技术来看,短读长测序提供了很高的z确性,但仅提供了少量数据片段,从而只能得到不完整的图片;而传统的长读长测序,可提供更大的图像,但缺乏z确性,因此很难分辨出真实的生物学变异与测序错误之间的区别。然而,兼顾长读长与高精度的HiFi测序正在改变一切,今天我们就来聊聊HiFi测序以及百迈客PacBio SequelⅡ平台HiFi产出情况吧。
一、何为 HiFi测序
HiFi reads(High Fidelity reads)是2019年由PacBio推出的基于环化共有序列(Circular Consensus Sequencing,CCS)模式产生的既兼顾长读长(10-20kb的长度)又具有高精度(>99%z确率)的测序结果。
在CCS测序模式下(图1),酶读长远大于插入片段长度,聚合酶会绕着模板进行滚环测序,插入片段会被多次测序。单次测序中产生的随机测序错误,通过环形测序生成的一系列Subreads来进行自我打磨,通过算法进行自我纠错校正,最终得到高z确度的HiFi reads。
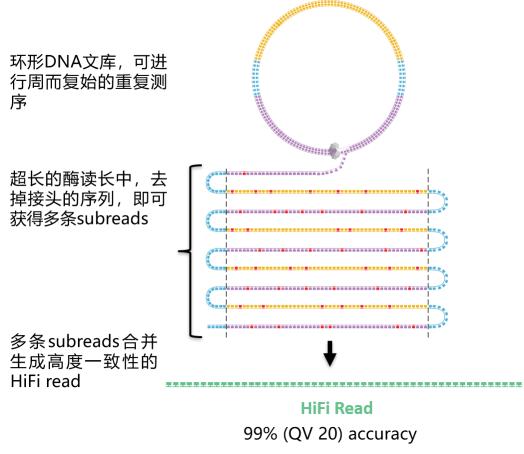
图1 HiFi reads是如何生成的
二、SMRTbell文库的构建流程简述
1.SMRTbell文库的结构
bell即“铃”的意思,如图2,构建完成的bell文库形状就如同一个哑铃。其主要组成部分是:发卡状的接头(Hairpin Adapter)和双链DNA模板(Double Stranded DNA Template)。而文构建完成后、测序前还需要完成bell文库、Sequencing Primer、DNA Polymerase的混合工作(测序引物退火结合bell文库,然后引物-bell文库复合物结合DNA聚合酶)。最终产物如图3所示。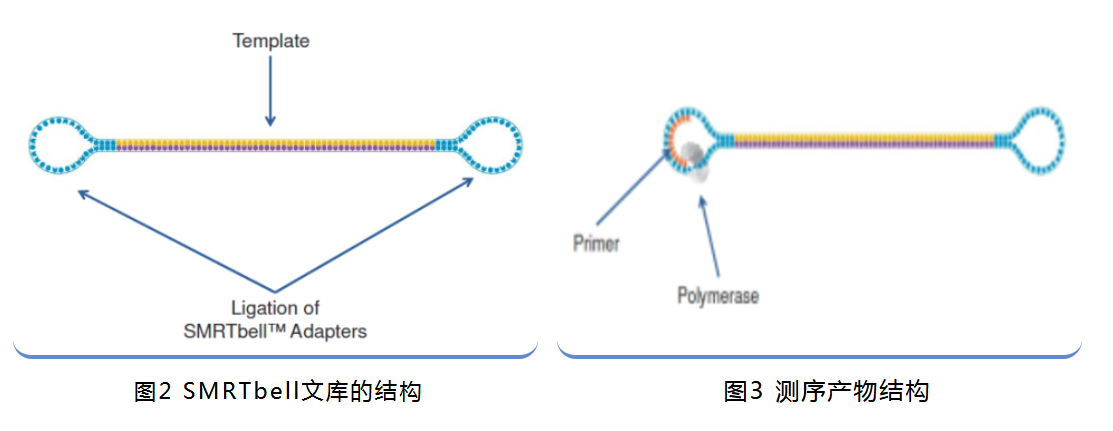
2.SMRTbell文库构建流程
以基因组HiFi文库为例(15-20K文库)(图4)。当得到gDNA后,先利用G-tube管或Megaruptor System将基因组片段化至合适大小,而后通过去除单链悬突、损伤修复和末端修复等步骤,得到完整的双链插入片段。接下来,通过将接头连接至双链DNA来创建SMRTbell文库,从而得到环状模板。完成接头连接后,需要对连接产物进行纯化,利用酶处理(图5)来消化线性或内部损伤环形DNA分子(游离的Hairpin Adapter、两端未连接Adapter的DNA模板、已成环但内部有损伤的DNA模板),酶处理完毕后,一般会利用Bulepippin或Sage ELF System切胶回收目标大小范围内的文库。
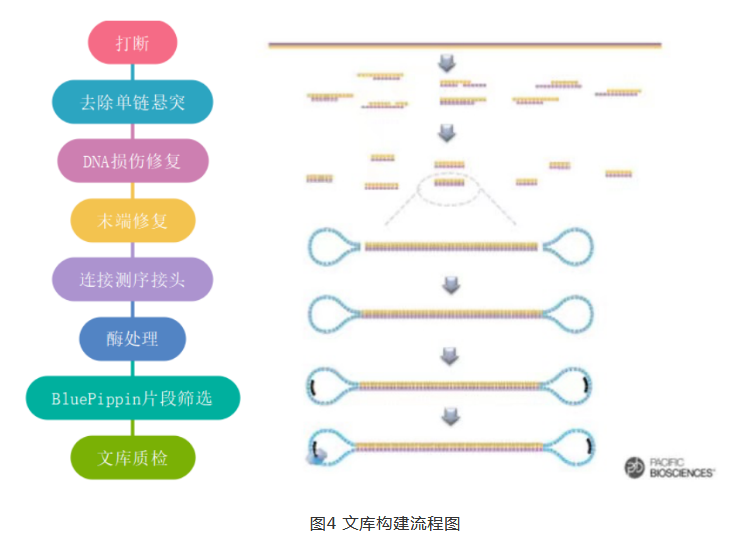
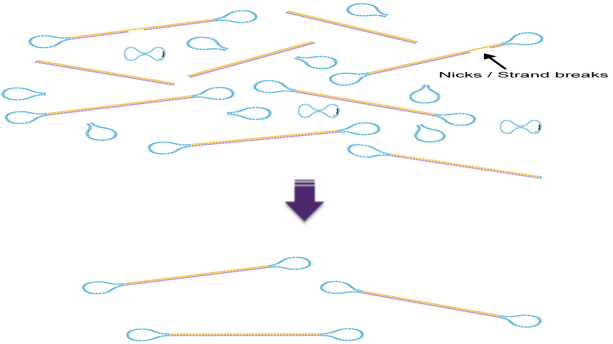
图5 酶处理示意图
三、HiFi测序的性能
1.使用HiFi Reads 进行基因组De Novo组装的能力
在基因组从头组装方面,研究者利用HiFi reads应用FALCON、Canu和wtdbg2算法分别对HG002基因组进行了从头组装,结果显示组装质量均较高,contigN50超过15Mb,并且与HG002标准结果高度一致,吻合率达到99.9983%(Q47.7)[1]。
表1 不同测序技术及分析流程组装结果

2.使用HiFi测序检测人类基因组变异的能力
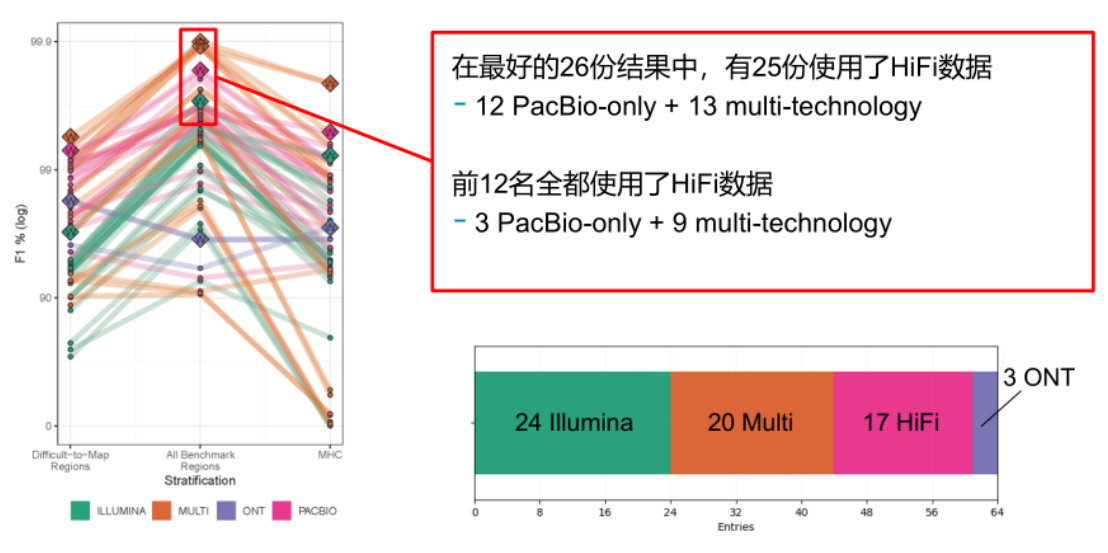
基因组测序中重要的自然是z确率,只有保证较高的z确率,基因组的研究才有价值。下图展示了最近的PrecisionFDA 真实挑战赛V2的结果(图6),在单一技术参赛结果中,使用PacBio HiFi数据(粉红色)在所有类别中,无论是全基因组范围(“所有基准区域”),还是在难以映射的区域或是主要的组织相容性复合体(MHC)中均提供了较高的z确性。所有的多技术参赛结果(橙色)中都使用了PacBio HiFi数据[2]。
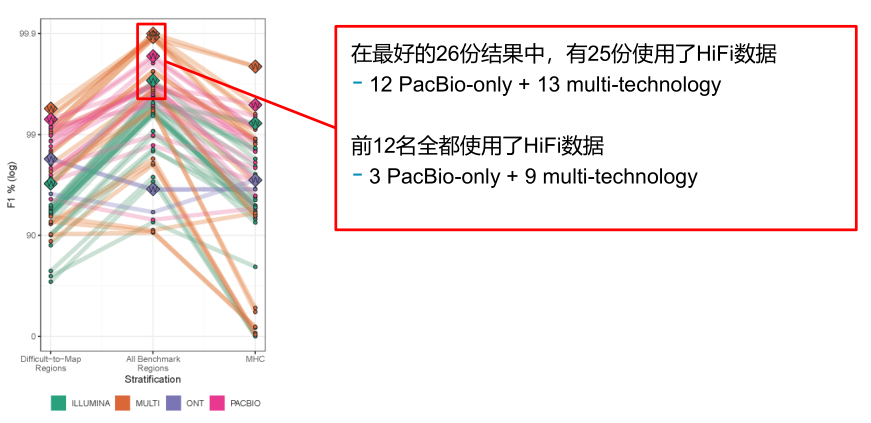
图6 PrecisionFDA Truth Challenge V2结果
另外,由下图可以看出(图7),Google DeepVariant使用HiFi数据提交的结果在所有单一技术检测全基因组范围内的变异z确性*高,对SNV√确度和召回率可以达到99.9%,对插入缺失的√确度和召回率可以达到99.4%[2]。
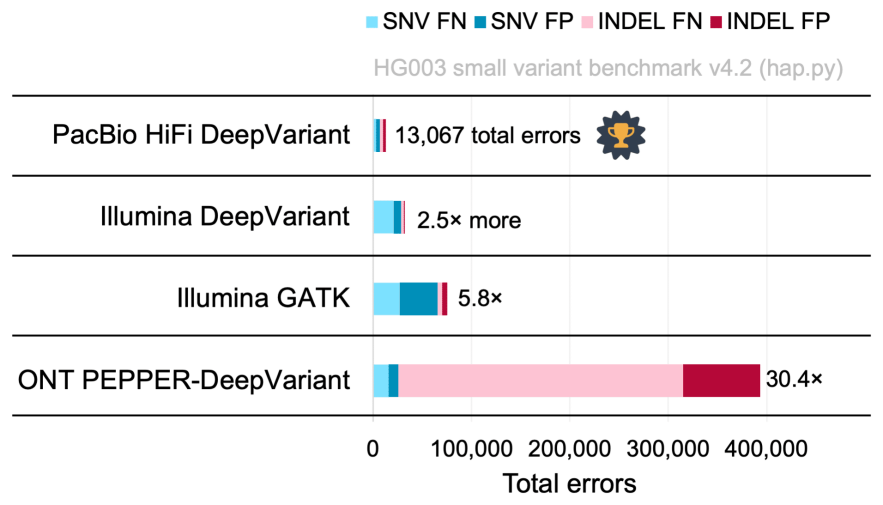
图7 不同测序技术及分析流程结果对比
四、百迈客HiFi测序数据展示
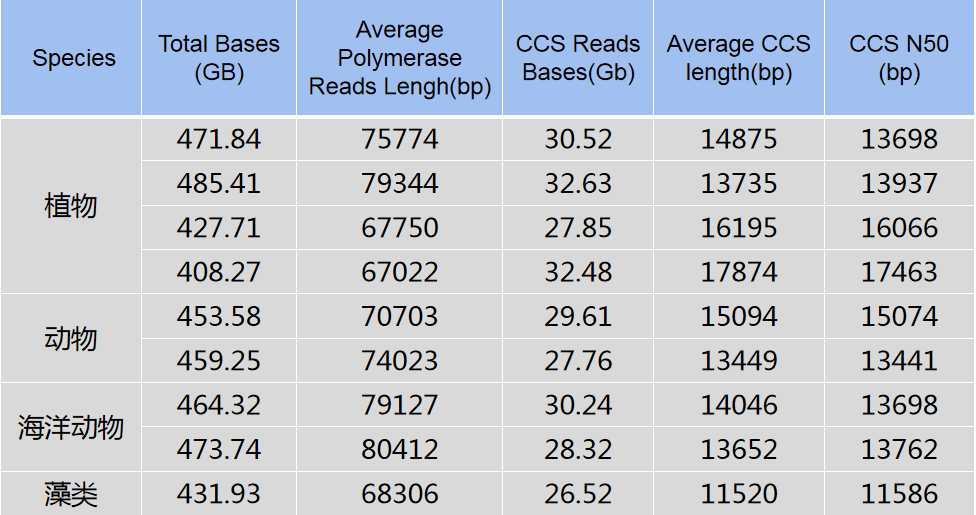
百迈客自2019年引进PacBio SequelⅡ平台以来,在HiFi测序方面已经积累了大量的经验,在技术人员的不断优化下,HiFi文库单cell产出更是有了新的突破,下面跟大家分享一下部分HiFi文库产出情况(表2)。在统计近1个月的HiFi cell中,我们单cell平均产出达416Gb。其中,单cell产出达400 Gb以上的占比达68%,同时,单cell的HiFi reads数据量高达32 Gb,占原始产出的比例*高可达7.96%。在读长方面,平均酶读长已超70Kb,HiFi reads长达18Kb。
表2 百迈客部分HiFi文库下机数据产出统计表
HiFi数据由于其长读长和高z确性,结合针对HiFi reads开发的组装软件,在基因组组装上有着较大优势。一般物种,单套30×CCS数据即可满足基因组组装需求,且无需繁琐的纠错过程,缩短组装时间,并能够识别复杂基因组区域的细微差别,有助于增加基因组组装的连续性、z确性和完整性。
在基因组组装方面,HiFi测序正受到众多科研工作者的青睐,已经成为越来越多研究者的不二之选,百迈客自2015年国内引进PacBio三代测序平台以来,在基因组研究领域已经有近百余篇合作文章发表于世界知名期刊,累计影响因子600+,目前已经拥有成熟的从测序到分析的完整HiFi流程,欢迎各位老师前来咨询!
参考文献
[1]Wenger A M , Peluso P , Rowell W J , et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome[J]. Nature Biotechnology, 2019, 37(11).
[2]PacBio.In precisionFDA Challenge,PacBio HiFi Reads Outperform Both Short Reads and Noisy Long Reads.https://www.pacb.com/blog/precisionfda-challenge/[EB/OL].2020.08.11




 京公网安备 11011302003368号
京公网安备 11011302003368号