
一个物种不仅需要高质量的基因组序列信息,同时还需要高准确的基因注释信息,这是后基因组时代功能基因组学研究的基础,因而进行高质量的基因注释显得尤为重要。
一、真核生物基因结构及注释方式
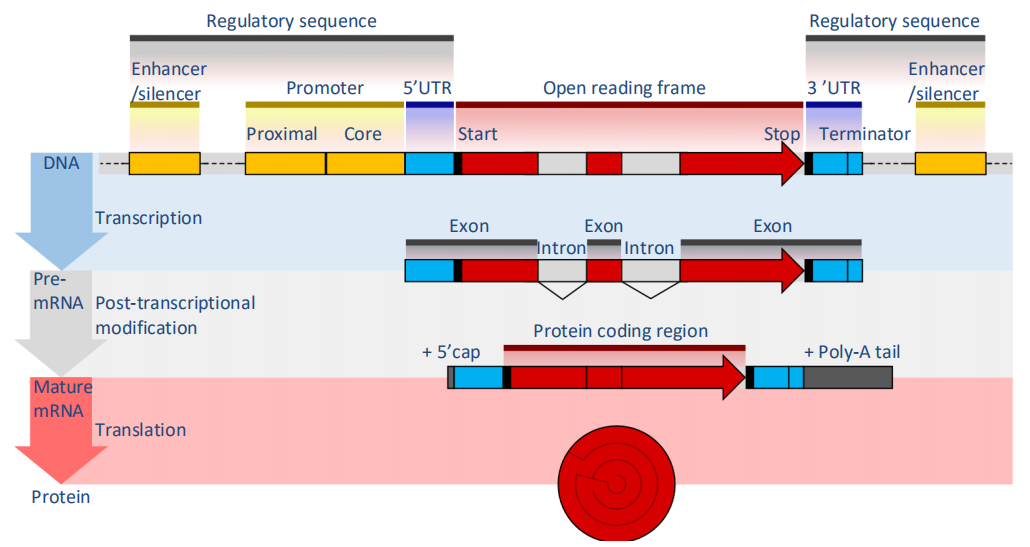
真核生物基因在结构分为外显子和内含子,在转录过程中会修剪内含子,并拼合外显子最后形成转录本。正是由于这种剪切形式的存在,造成了真核生物基因可以采用不同的剪接形式(可变剪接),形成不同的转录本,从而发挥更加广泛且精准的作用,这也导致了真核生物基因结构注释难度较大。另外基因组中大量非编码区域的存在也严重影响基因的定位,造成精准注释的困难!
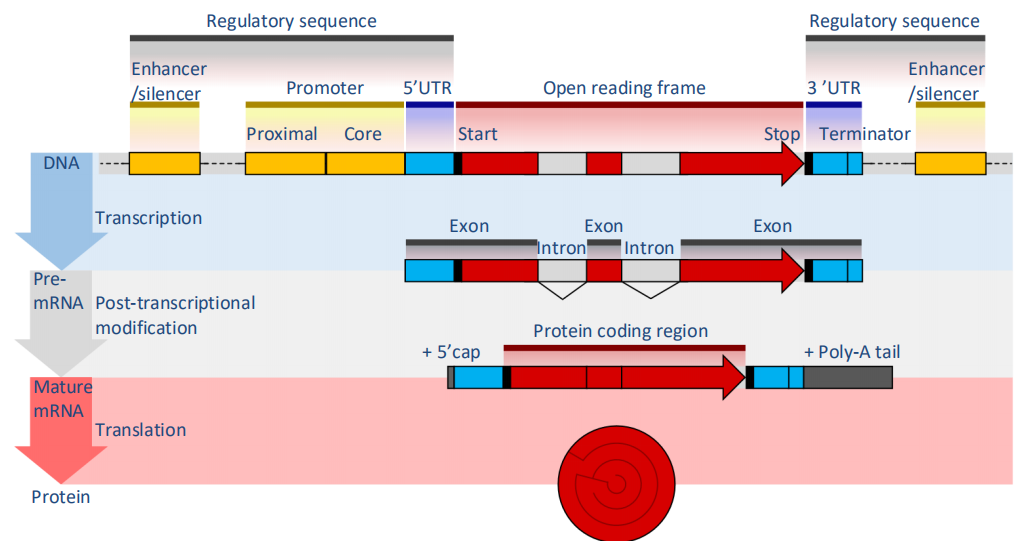
图1 真核生物基因结构[1]
(1)同源预测(homology-based prediction):目前已发表大量基因组,可以利用物种间基因序列较好的保守性,使用已有的高质量近缘物种注释信息通过序列联配的方式确定外显子边界和剪切位点。目前常用的软件有GeneWise、GeMoMa等,GeMoMa预测效果较好。
(2)从头预测(de novo prediction):通过已有的基因特征训练模型来预测基因结构,一般预测基因数量较多,准确性较低,但在有好的训练集条件下,如有全长转录本提供训练集和预测证据,也能取得很好的效果。常用的软件有AUGUSTUS、GlimmerHMM、SNAP、GeneID、GenCsan等。
(3)基于转录组预测(transcriptome-based prediction):指通过物种各个组织混合的RNA-seq和三代全长转录本数据数据来辅助预测。由于转录组这种数据是转录本结构的直接反应,因而利用此种类型数据,可以比较真实准确的确定外显子区域和剪切位点,在基因预测三种策略中属于可靠性最高的策略。常用的软件有PASA、TransDecoder、GeneMarkS-T等。
总之三种方法各有优劣,如同源预测受限于近缘物种注释质量,但在近缘物种注释质量较高的前提下可以实现大多数基因的准确注释;从头预测,预测基因数量多,但完整度好,不容易丢失一些基因,尤其是在近缘物种中没有的新基因,但其不可避免引入一些假的预测结果;转录组预测,准确度最高,但由于表达的基因一般也就只有60%~70%左右,所以检测到的基因数量有限。故最后一般要用EvidenceModeler(EVM)等工具进行三种预测策略的整合,再进行后续的蛋白功能域、代谢通路等的注释。
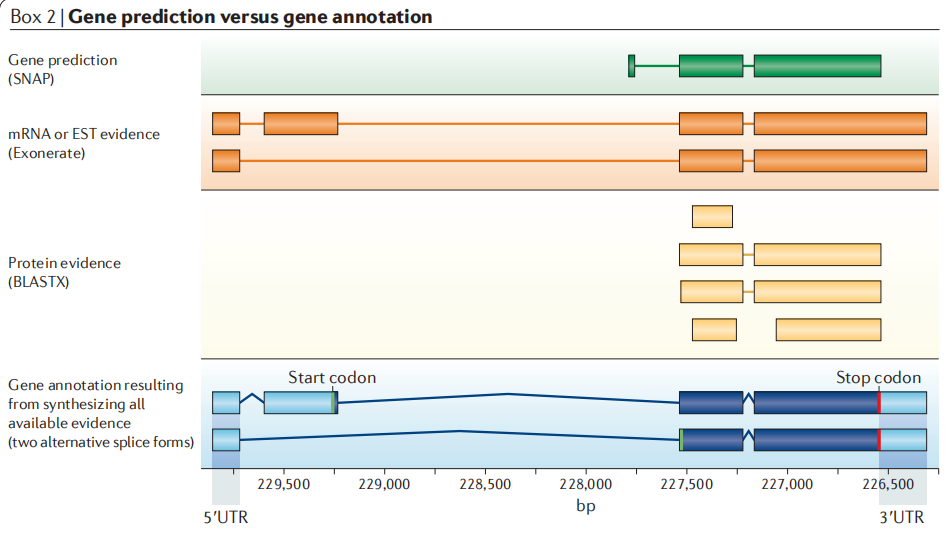
图2 从头预测、基于转录组预测及同源预测基因结构展示[2]
二、二代转录组和三代全长转录组在基因预测中对比
二代转录组测序技术,通常打断成小片段进行测序,后期通过对小片段组装(如采用Tirnity软件),获取相对完整的转录本。但是由于测序片段较短可能会存在组装错误或者组装不完整,导致不能准确获得完整转录本,进而对基因预测的完整性和准确性产生严重的影响。
01 获得准确的完整转录本
基于三代测序平台可以直接获取转录本的5ˊ到3ˊ高质量全长序列,无需组装,一条read即可跨越全长转录本,因此通过将read比对基因组就能够非常容易的确定基因在基因组上的位置和其完整结构,因而非常有利于基因的注释工作,且准确性较高(参见下图以水稻一个基因为例)。
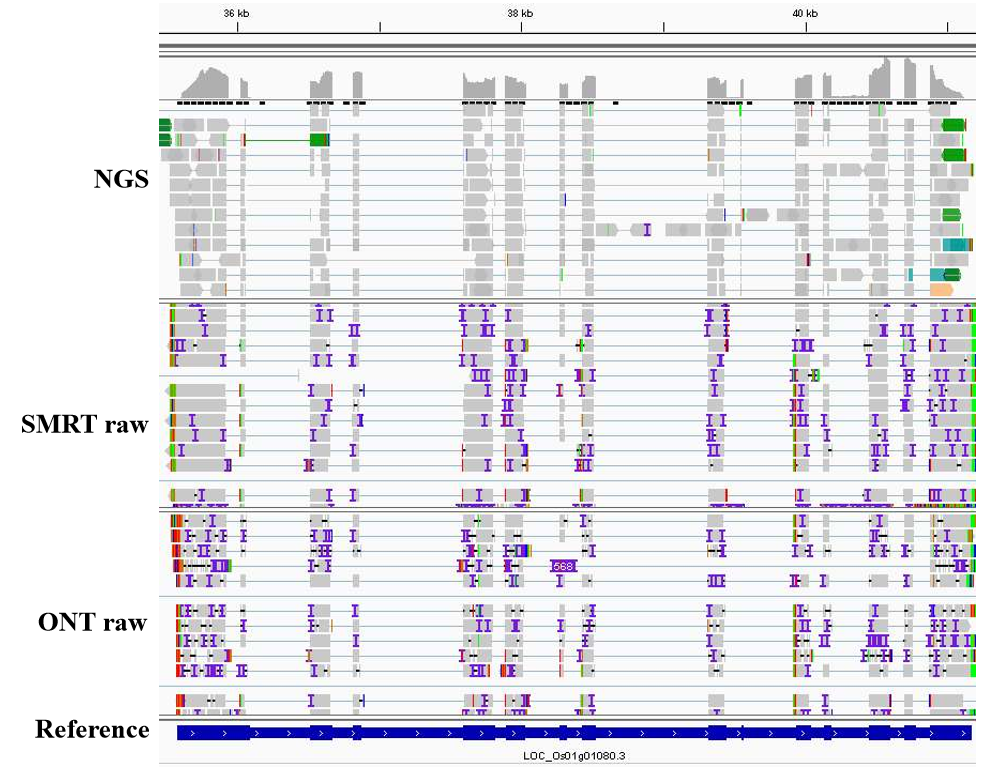
图3 三种数据比对基因组结果
图3中最下方为参考转录本结构,可见三代平台均实现一条read覆盖,且存在多条read同时支持,二代平台则由众多短read组成,后续组装需借助算法才能组装到完整转录本,存在不确定性。
02获得准确的可变剪接
在可变剪接方面,三代全长测序结果可以捕获更多、更准的、不同可变剪切形成的转录本,如图4显示,Exon2和Exon6以及Exon9是三个转录本间共享,二代多数短read单条连一个外显子区都无法跨越,对于完全比对到这3个外显子区的短reads无法区分其来源转录本。而Nanopore长读长测序可以直接得到3种全长转录本,因此对于转录本可变剪接识别更准确。
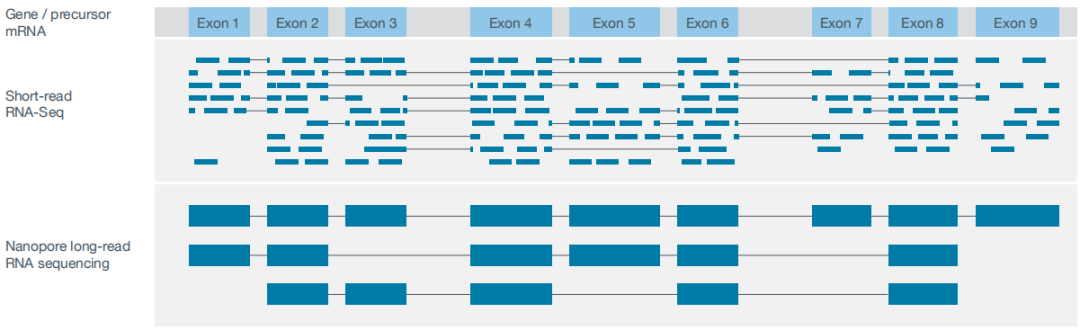
图4 二代转录组与ONT全长转录组识别可变剪接
三、Nanopore在三代全长转录组辅助基因预测中的优势
01数据及成本更亲和
长度长在转录本分析方面比短读长测序技术有明显的优势。PacBio平台由于测序原理限制,一个ZMW孔只能循环测序一个分子,加上芯片中 ZMW 孔数限制,需要较大的数据量以及更多的测序芯片才能达到饱和,成本相对较高。Nanopore测序原理使得在一个分子穿越纳米孔后其他分子还可继续穿行,更少的数据量可以获得更多的信息,因此成本也更加亲和。百迈客研发团队将相同物种不同平台的结果进行比较发现,当预测到数量近似相等的高准确的基因时,PacBio平台所需转录组数据量远远多于Nanopore(表1)。
表1 不同物种PacBio与Nanopore获取相近高准确基因数量时对应所需数据量
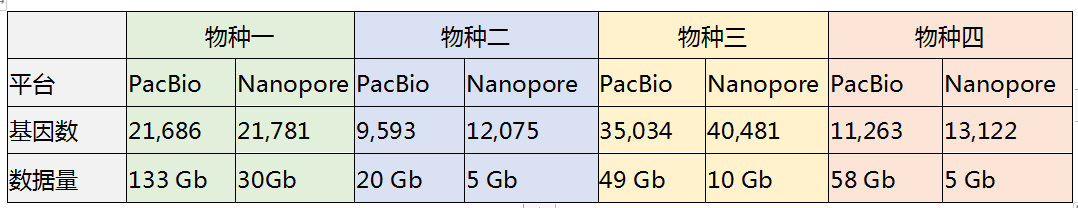
注:基因数:identity和coverage均大于90时的高准确基因数
02准确性具有保障
三代测序的错误率太高,会不会对结果有影响呢?目前Nanopore下机数据准确率已经可以到90%,即碱基平均错误率为10^(-1)=10%左右,完全可以将read准确的回帖到参考基因组中的,因此不会出现错误比对的情况。唯一需要解决的是可变剪接位点比对位置的准确性,目前我们基于自己开发的软件NanoGAP,借助于二代RNA-seq数据、自身ONT数据及其他方法预测结果,共同纠正转录本剪接位点,实现了在小数据量情况下与PB CCS模式下同等的准确性(见表1及图5)!
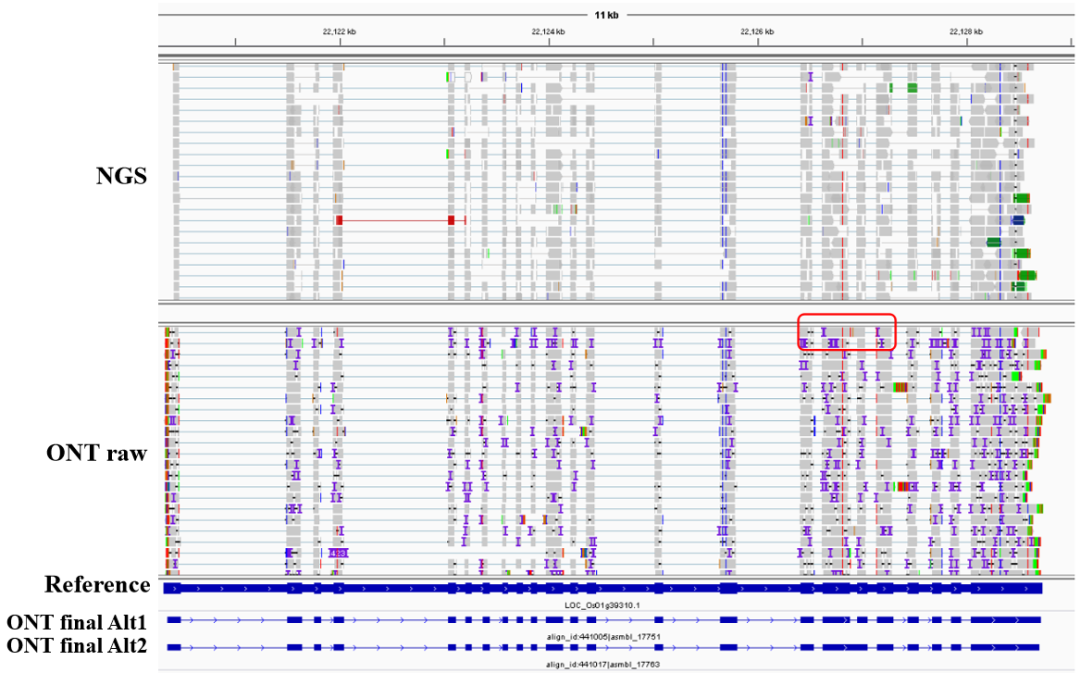
图5 NanoGAP预测结果示意图
图5中分别展示了RNA-seq和ONT原始数据比对结果,参考基因的结构和经过NanoGAP纠正和聚类后此位置转录本的结构。其中NanoGAP预测到的ONT final Alt1转录本,其结构与参考基因结构完全一样,证明我们采ONT数据进行预测的高准确性。同时NanoGAP预测到的一个新的可变剪接ONT final Alt2,表明拥有更长读长的ONT在获取可变剪接方面存在一定优势。
03物种与数据量需求
进行基因的预测,不同于有参条件下进行基因的表达定量。由于三代测序存在一定的错误率,所以我们需要通过增加数据量实现对转录本的纠正,进而获得高质量的转录本,用于基因的辅助预测。那么多少数据量适合呢?不同的物种的需求是否不同呢?百迈客研发团队抱着这样的疑问进行了多轮多物种Nanopore数据检测,研究结果显示不同物种具有数据量需求差异,在5-20 Gb时随着数据量的增加,预测到的高准确基因数迅速上升,部分物种在10G左右预测到的高准确基因数量可以达到最大,在数据到20 Gb左右时,绝大多数物种已达到高准确基因覆盖度饱和。而多倍体物种在20-30 Gb时,高准确基因覆盖度基本达到最大。
04具有拓展应用
组织差异越大,基因的表达差异通常也越大,因此基于转录组预测的方式需要多组织进行混样分析以获得更全面的基因信息。与PB相比ONT还具有无GC含量和碱基偏好性、转录本表达定量准确的特性,在混合数据应用于注释分析前,可以用作多组织/多处理下的表达差异分析。实现样本一次检测多种分析,完成数据最大利用率,也为文章添光增彩。
四、案例分析
研究者分别用二代与三代全长转录测序对锡兰钩口线虫(Ancylostoma ceylanicum)基因结构预测,研究发现三代全长测序与二代测序相比的一个显著特点是UTR的数量和长度增加,尤其是3’UTR,带有3’UTR和5’UTR的基因数量分别增加了5倍和3倍。研究结果说明了长读长在定义基因UTR和因此更完整的ORF方面的优势。而研究表明UTR区域与真核生物中基因表达调控的复杂性相关,进一步说明了全长测序在基因发现和识别基因边界方面的重要性[3]。
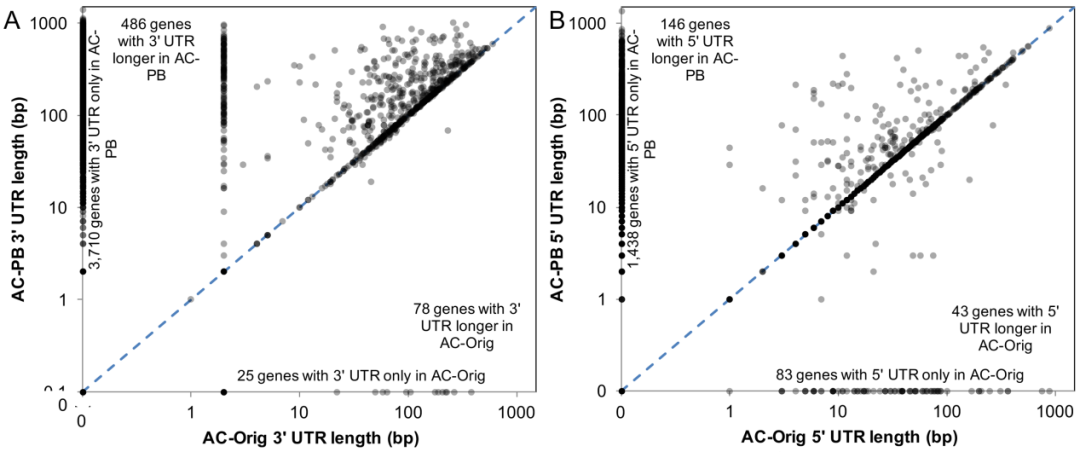
图6 (A)3’UTR和(B)5’UTR的全长和RNA-seq之间UTR长度的差异
进一步的基因鉴定结果显示,基于二代转录本的BLASTX(红色)和protein2genome(蓝色)预测了一个短基因模型,但是全长转录序列(绿色)扩展了现有的基因,并预测了一个新的基因[3]。除此之外,研究者通过三代全长技术共鉴定1609个(9.2%)新基因,表明了全长转录组的加入使基因注释更丰富。
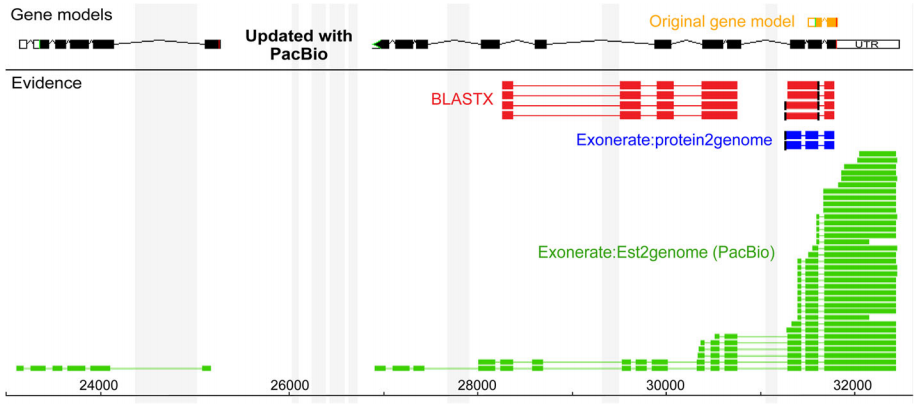
图7 二代与三代基因结构预测结果比较
五、尾声
综上所述,我们不难发现:全长转录组辅助基因预测与二代技术相比具有
①三代技术直接得到全长转录本,无需组装,结果更可靠,基因定位和结构注释更加准确;
②转录本可变剪接识别更加容易,结果也更加可靠
③转录本的5’和3’端覆盖更均匀、完整,基因的UTR(非翻译区)定位更加精准等优势;
而三代测序中,Nanopore与PacBio相比,又具有低数据量饱和的优势(大多数物种20 Gb基本可以达到饱和及分析需求),低成本高收益的方式为科研之路提供了新的方向。
(1)全长转录组提升玉米基因组注释(Li C et al., Nature Communications.2020):

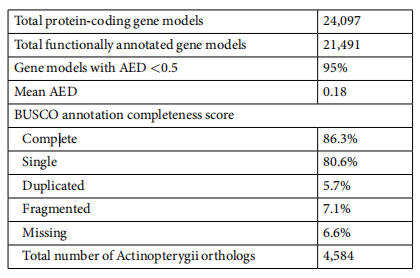
(2)ONT辅助金线鲻鱼转录组注释统计(Kadobianskyi M et al., Scientific Data.2019):
如果您对全长转录组测序技术感兴趣,您可以点击下方按钮联系我们,我们将免费为您设计文章思路方案。

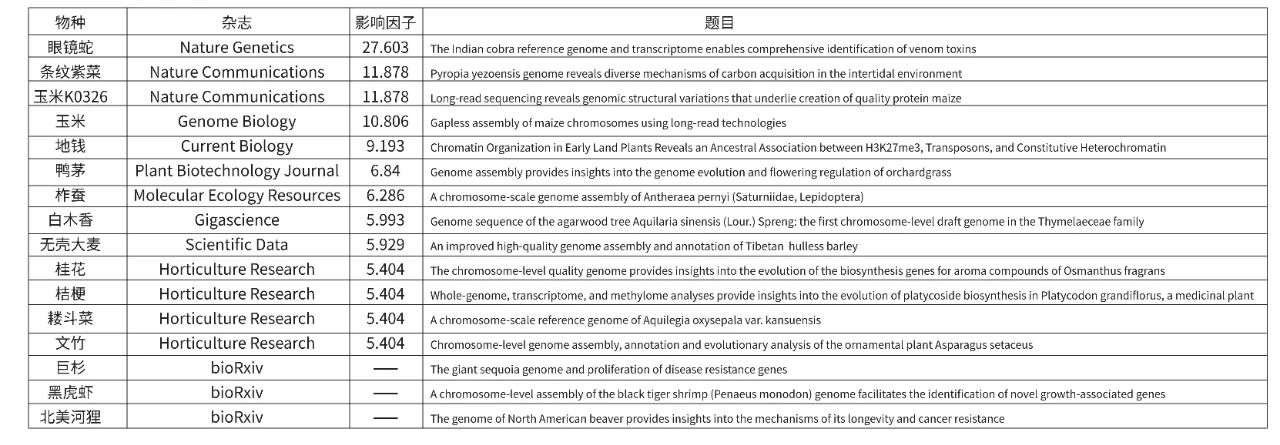
更多使用全长转录进行基因组注释优秀案例(2020):




 京公网安备 11011302003368号
京公网安备 11011302003368号