BSA(bulked segregant analysis)又叫集群分离分析法,该方法主要是选择双亲群体分离后代中具有极端表型的个体进行混样,通过比较不同极端混样池之间的多态性标记的差异,从而筛选出与性状相关的分子标记,实现目标基因的定位。BSA分析早期使用的都是传统分子标记,根据标记与性状共分离(紧密连锁)得到定位区间,不过由于其标记密度低、定位精度差、实验操作繁琐已经逐渐不再使用。近年来,BSA方法和高通量测序相结合,通过比较不同混池间SNP的频率差异或者比较混池间纯合SNP的密度的差异,实现基因的定位,高通量测序方式以其简单、快速、准确、高性价比的特点受到科研工作者的青睐。那么,对于不了解BSA的研究者来说,如何快速了解BSA的发展历程、原理方法、应用领域,如何快速上手BSA分析,今天大师兄为大家推荐几篇BSA代表性文章,让你轻松搞定BSA,赶紧上车。
BSA的提出
文章题目:Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations
发表期刊:PNAS
发表年份:1991
文章要点:
(1)在这篇文章中BSA方法被首次提出来,是BSA的开山鼻祖。
(2)该文章利用传统的RAPD标记,首先通过筛选亲本间多态标记,然后在性状分离的杂交群体中选择性状极端个体组成混池,根据多态标记与混池性状共分离的原理,获得与性状连锁的markers。
(3)利用该方法,作者对莴苣霜霉病抗性基因进行的了定位。
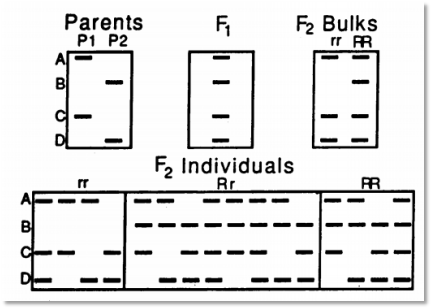
图1 BSA方法筛选共分离标记
BSA的进阶
1、文章题目:Genome sequencing reveals agronomically important loci in rice using MutMap
发表期刊:Nature Biotechnology
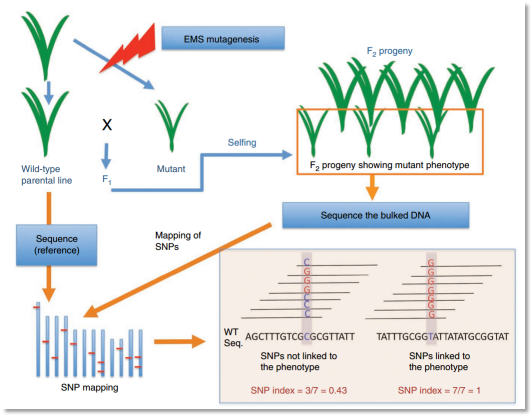
(1)该文章提出了一种针对化学诱变剂的点突变性状的定位方法—mutmap,该方法是目前在做植物诱变性状研究的主要方法。
(2)该方法使用突变个体与其野生型个性进行杂交得到F2群体,在群体中选择突变表型的子代组成1个突变混池,然后只对混池DNA进行高通量测序。
(3)该方法中首次提出了SNP-index的概念,SNP-index是目前最常用的BSA定位方法之一,即计算混池中非参考基因组类型的SNP的频率,同时再利用滑窗法对连续多个SNP的SNP-index值进行拟合,最终获得性状关联的基因组区间。
(4)作者利用该方法对水稻夜色突变基因进行了定位,并且在chr12的尾部定位一个候选基因OsCAO1。
图2 mutmap定位原理示意图
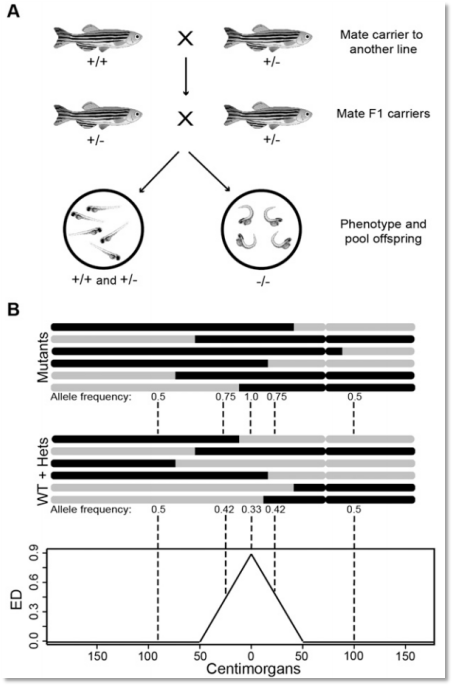
2、文章题目:MMAPPR: Mutation Mapping Analysis Pipeline for Pooled RNA-seq
(1)该文章首次提出了MMAPPR的方法对突变性状进行基因定位,该方法最大的亮点是对极端个体混池的RNA进行测序,而不再是混池的DNA进行重测序,这对于基因组较大的物种来说无疑不是一种最佳选择。
(2)对于没有亲本的杂交子代群体,首次将欧式距离算法(Euclidean distance,ED)应用到性状关联分析中,同时提出通过ED值乘幂的方式实现背景噪音的过滤。
(3)在性状关联时,使用了LOESS拟合的方法对ED乘幂值进行拟合,获得显著关联的基因组区间。
(4)利用该方法,作者对斑马鱼心血管突变体性状进行了分析,最终定位到2个候选基因ctr9和cds2。
图3 MMAPPR的定位过程
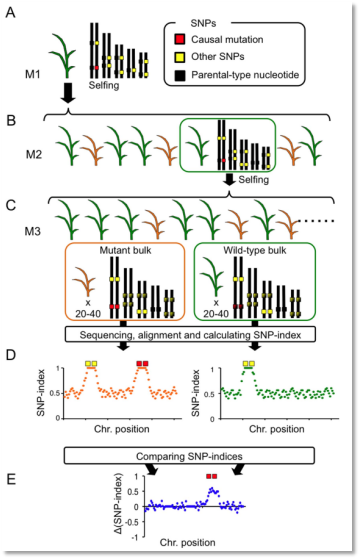
3、文章题目:MutMap+: Genetic Mapping and Mutant Identification without Crossing in Rice
(1)该文章是对突变性状定位的一种延伸,也是对mutmap方法的一个补充,主要区别是如果隐形突变个体无法正常发育在幼苗期出现死亡,因此无法与野生亲本杂交构建分离群体,所以该方法巧妙的选择了突变位点杂合的个体进行自交,从而在自交M3或者M4选择极端个体构建混池进行后续分析。
(2)该方法为了避免SNP在自交过程中的固定效应和群体中的偏分离标记,不再像mutnap分析中对一个突变性状混池进行测序,而是对2个极端混池都进行测序,从而降低了背景噪音。
(3)在SNP-index关联算法的基础上,进一步发展提出△SNP-index算法,通过△SNP-index的大小判断性状关联区域。
(4)利用该方法对水稻浅绿叶色突变进行了分析,定位并验证了OsNAP6基因的功能。
图4 MutMap+的定位过程
4、文章题目:QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations
(1)前期的BSA定位方法主要集中在发生突变的质量性状上,该文章提出了针对植物数量性状的研究方法QTL-seq。如果一个性状是由多基因调控,分离群体子代的性状呈现正态分布,QTL-seq方法选择极端表型的部分子代构建混池并进行高通量测序,该方法是目前数量性状定位的主流方法。
(2)对于SNP-index关联值提出了置信区间的算法,将定位结果锁定在基因组某个区间范围内。
(3)利用该方法作者对水稻稻瘟病菌抗性进行了定位,并且在chr6头部定位到一个显著的关联峰。
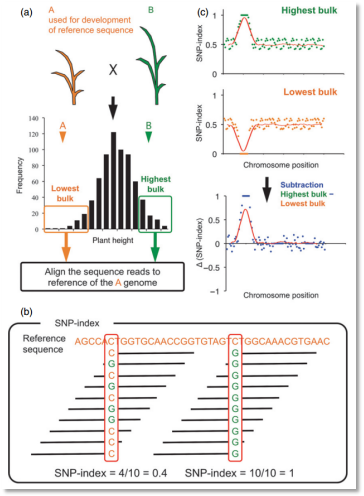
图5 QTL-seq的定位过程
BSA的升级
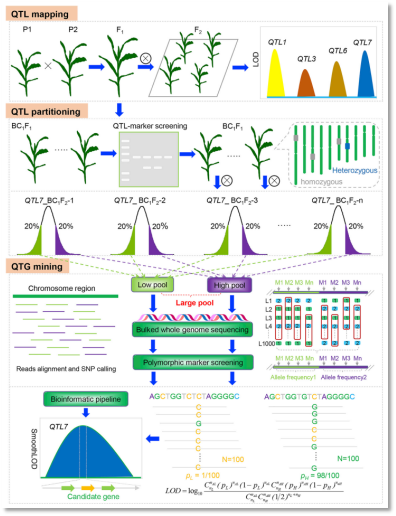
1、文章题目:QTG-Seq Accelerates QTL Fine Mapping through QTL Partitioning and Whole-Genome Sequencing of Bulked Segregant Samples
(1)该方法是整合了遗传图谱QTL定位、回交子代背景选择、QTL-seq于一体的综合方案,项目开展前需要经过精心的试验设计,该方法可以实现数量性状或者质量性状调控基因的精细定位,并且可以对某个性状的多个主效位点同时进行定位。
(2)首先通过F2代遗传图谱进行初步定位,获得某个性状所有QTL信息,然后从BC1F1中筛选目标QTL杂合,其它背景QTL纯合的单株,自交一代得到BC2F1,从大规模BC2F1再选择极端性状个体组成混池进行QTL-seq,去进一步缩小定位区间。
(3)作者以玉米株高为模式性状展开QTG-seq研究,在chr7上关联到候选基因Zm00001d020874。
图6 QTG定位研究思路图
2、文章题目:Dissecting a heterotic gene through GradedPool-Seq mapping informs a rice-improvement strategy
发表期刊:Nature Communications
(1)普通QTL-seq选择分离群体中极端性状的子代个体构建2个混池,而GradedPool-Seq不仅选择了极端个体,还把中间表型个体加入其中构成1-2个混池,最终构成“高值组”,“中值组”,“低值组”,再进行QTL的定位。相比之下,GradedPool-seq的定位精度通常要优于仅使用两个极端池进行的常规BSA。
(2)不再使用常见的SNP-index和ED算法进行性状关联,而是采用Ridit(relative to an identified distribution unit)算法会对每个位点的等位基因频率进行计算,再对计算得到的p值进行滑窗法拟合,从而显著降低背景噪音,实现精细定位。
总结
以上都是BSA方法必读的经典文章,从传统的BSA分析筛选共分离markers,到BSA与高通量测序相结合的精准定位策略,极大地拓展了基因定位方法的范围,能够让你从原理到方法全方面的了解BSA,如果后续开展这方面的研究,这些文章将会促进你快速入门。如果还有什么疑惑,欢迎点击下方按钮联系我们,我们可以为您免费进行文章研究思路设计。
当然,在了解BSA理论方法的同时,亲自动手分析也是不可或缺的。可能有些研究者就会困惑了,自己没有生信基础,不会敲代码也不会跑流程,更没有服务器等硬件设备,自己怎么分析BSA数据呢?交给服务公司是个不错的选择,不过项目成本就会增加,而且项目周期一般较长。那么,有没有一种方式,可以让研究者既不需要生信基础,同时降低项目成本和缩短项目周期呢?
当然有了,那就是百迈客云BSA分析平台。该平台整合了目前最主流的BSA分析流程,定位结果准确有保证;分析过程简单易上手,从数据上传到参数设置,再到基因组选择,只需要简单3步就可以完成项目数据投递和分析;结题报告云交付,分析完成即可拿到项目结题报告;多种常见定制化分析,助力BSA数据深入挖掘(开发中)。






 京公网安备 11011302003368号
京公网安备 11011302003368号