
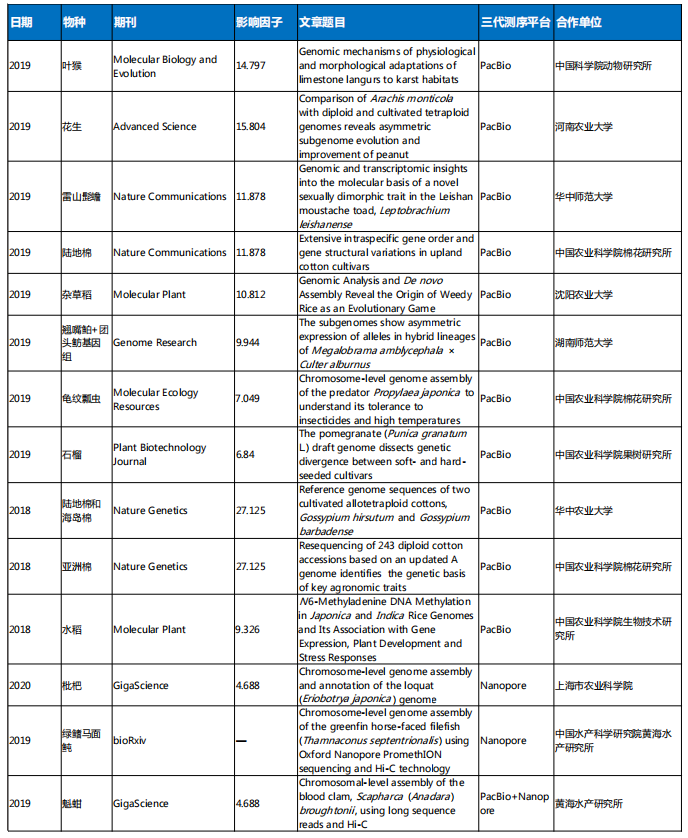
DNA测序技术在过去的40年中,经历了巨大的改进与变化。早在1977年,首次报道了Sanger和Maxam–Gilbert测序方法,Sanger测序的最大序列长度约1 kb。其对DNA总量的要求较高,一般通过克隆靶标DNA序列并连接载体,进而通过原核细胞大肠杆菌(E. coli)扩增(当时基因组De novo采用BAC文库测序方式),其读长短且耗时;NGS(Next-Generation Sequencing )二代测序包含很多技术平台,其特征是对大量的DNA分子并行测序,多年来已有4个主要的NGS平台投入商业使用:罗氏454平台, Illumina GA/Solexa 平台, ABI SOLiD平台和Life Torrent平台。在过去的10年中,Illumina因其低成本,高速和高产而成为测序市场的主要供应商,Illumina测序平台具有广适性,因此NGS已广泛用于探索基因组学的各个领域,包括肿瘤学,微生物学,环境基因组学,宏基因组学及医学,环境和农业研究等,随时其广泛的应用,其劣势也逐渐的突显,即:二代测序(Illumina为代表)读长短仍然是生物学研究的重要瓶颈,这限制了许多生物学研究的准确性,尤其是在基因组组装研究中。在片段重复(segmental duplication),结构变异(SV,structural variations)或旁系同源区段分析中使用短读长测序可能会导致大量假阳性。尽管测序技术和生物信息学分析在进步,但大型基因组的从头组装仍然具有挑战性。自2015年起,以PacBio和Nanopore为代表的长读长测序技术开始在动植物基因组De novo中初露锋芒(图1 A和B)。

图1 不同测序技术读长,准确性及基因组连续性评估
一、三代长读长单分子测序技术的发展
长读长单分子测序技术(Long read single-molecule sequencing technology)又称第三代测序技术TGS(Third-Generation Sequencing),早在2004年,由美国太平洋生物科学公司Pacific Biosciences (PacBio) 创立的实时(SMRT)测序是较早被广泛使用的长读测序技术,SMRT测序产生的Reads可达到约200 kb。其提供了技术上的优势,以鉴定遗传变异并进一步研究其基因功能,同时作为动植物基因组组装日臻进步完善的主要驱动力,自2015年,首篇纯PacBio三代数据组装复活草(Nature. 2015)基因组见刊Nature,开启了三代动植物基因组De novo的纪元。与Sanger测序和NGS测序类似,PacBio测序同样采用边合成边测序的方式,以其中一条DNA链为模板,通过DNA聚合酶合成另外一条链(图2 A和B)。PacBio测序平台相继推出RS II,Sequel和Sequel II平台并投入使用(Table 1)。2005年,英国牛津纳米孔技术公司
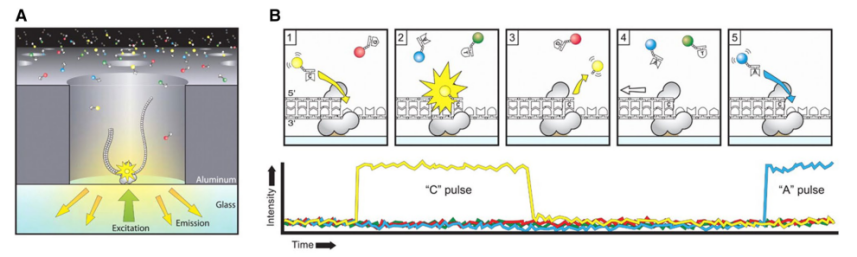
图2 三代PacBio测序原理
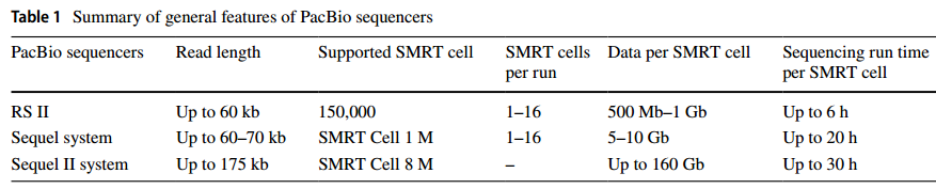
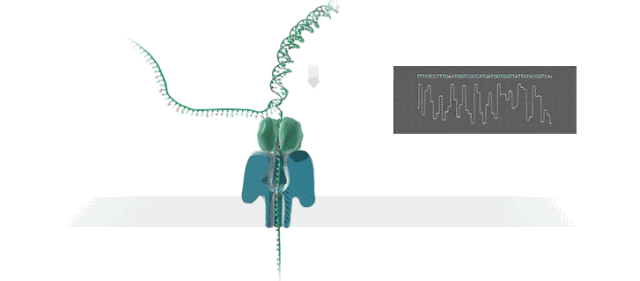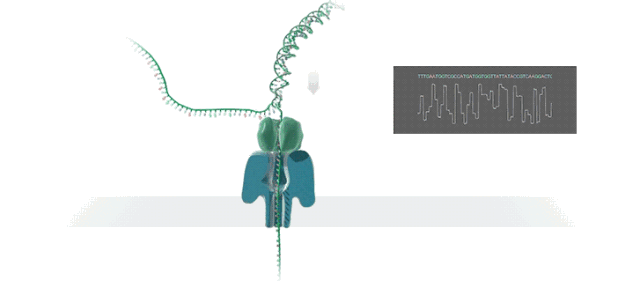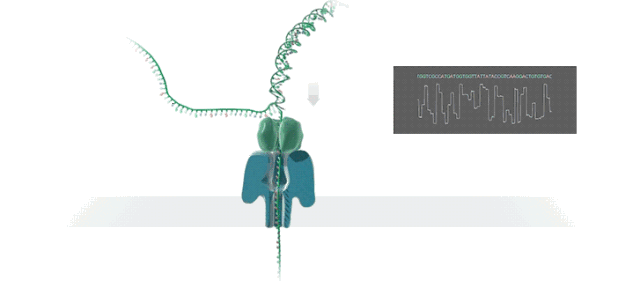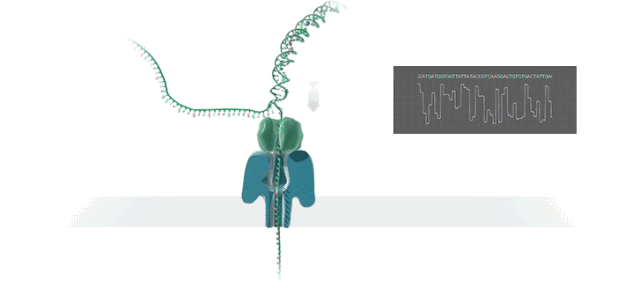
图3 三代Nanopore测序原理
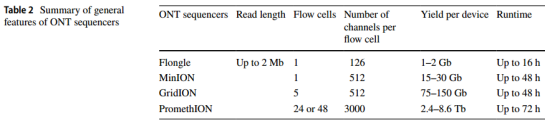
二、三代长读长单分子测序技术PacBio和Nanopore的比较
PacBio和Nanopore具有共同的优点,即长读长;同时也具有共同的缺点即高错误率(纠错前随机分布的〜5–20%碱基错误率),随着新测序仪和生物信息学的不断发展,测序平台的优缺点有望发生改变,无论是PacBio还是ONT测序平台都致力于获得更长读长的reads的同时,兼获高准确的碱基序列信息。
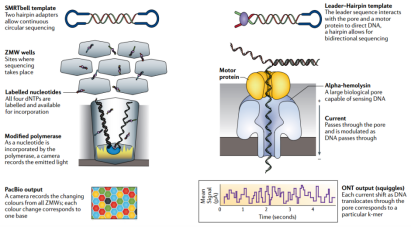
图4 PacBio与Nanopore测序原理及信号识别原理比较
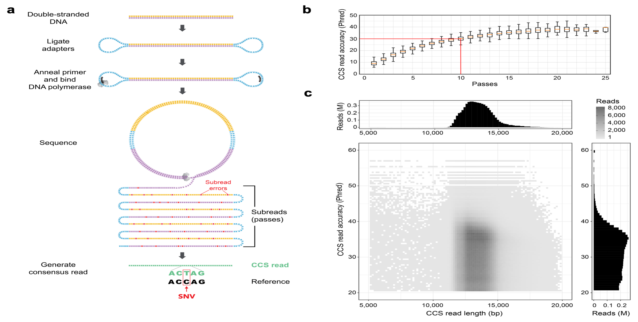
图5 PacBio CCS测序原理及准确性评估
Nanopore超长读长测序:尽管组装方法不断在改进,且已开发物理图谱技术(光学图谱),但读长长短仍然是高质量动植物基因组的限制因素。如植物基因组由于高杂合,及其复杂的多倍性和高重复含量,其组装仍然具有挑战性,读长必须超过基因组中的主要重复序列长度,及嵌合的长末端重复序列(LTR)或单倍型Blocks,其长度可能跨越20–200 kb。虽然PacBio是提供Long Reads(>1 kb)的技术,且通常 Reads N50长度可大于20 kb,但即便是几乎完美的15 kb reads可能无法组装复杂植物基因组中经常出现的嵌合及高度相似的重复序列。而ONT测序平台大大解决了这一问题,与PacBio reads平均长度项目(图6),一小部分ONT reads读长超过300 kb,同时PacBio不包含任何大于150 kb的reads。许多复杂的植物基因组具有大于20 kb或更长的重复序列,所以即便目前ONT具有一定错误率,但其大大促进了基因组的组装,从而显着提高了基因组连续性或完整性。例如:使用ONT测序更新的拟南芥Col-0基因组最终通过组装,减少到40个Contigs,且跨越了染色体臂(端粒到着丝粒),同时解决了前期在TAIR10参考基因组中存在的gaps及组装错误(Jupe et al. 2020)。
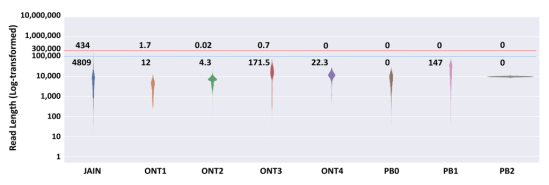
图6 三代Nanopore和PacBio测序读长比较
三、百迈客双平台(Nanopore+PacBio)动植物基因组De novo研究策略—鱼和熊掌可兼得
“鱼,我所欲也,熊掌亦我所欲也;二者不可得兼,舍鱼而取熊掌者也。正如在动植物基因组研究中,针对基因组组装,为了兼顾长读长的同时,获得高准确性的物种基因组密码信息,在选择测序技术选择(PacBio or Nanopore?)上总会有鱼和熊掌不可兼得的感觉。长久以来,百迈客一直致力于成为“专业的基因组组装专家”,拥有双平台的基础上(2015年首次引进PacBio平台;2017年首次引进Nanopore平台),力求整合双平台各自的优势,着力于开发各种软件、算法,为每个物种提供订制的“基因组套餐”,即打造高质量,高完整性的物种基因组。从本章节起,小编后续会结合新的技术策略、测试数据及文章案例,为大家带来全新的基因组研究策略,旨在获得高度连续性基因组的前提下,同时完成高准确性动植物基因组密码的破译,即鱼与熊掌可兼得。
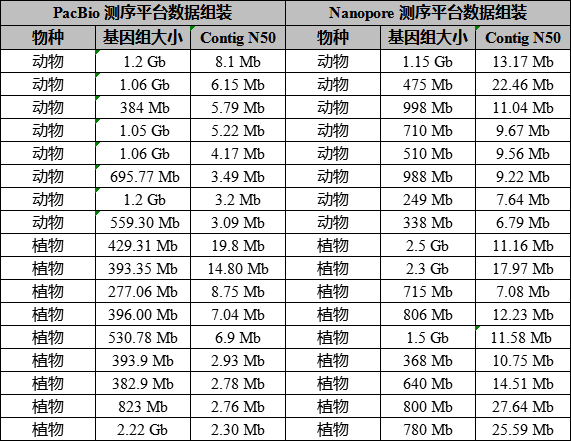
首先通过百迈客三代Nanopore和PacBio平台相关物种测序读长(表1)及组装结果的比较(表2),进一步通过我们的实际案例来看一下Nanopore测序平台在基因组组装中的优势。
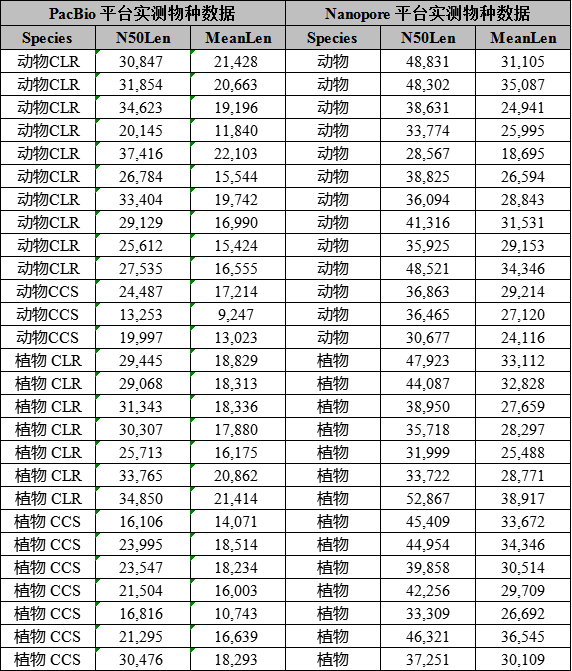
通过双平台实测数据的比较分析: Nanopore平台平均读长为28.5 Kb左右,Reads N50平均读长 38Kb左右;PacBio CLR平均读长20 Kb左右,Reads N50平均读长 28Kb左右;CCS平均读长12-15 Kb,Reads N50 16~18Kb,发现Nanopore比PacBio平台读长高10 Kb左右,而PacBio CCS模式读长远低于CLR模式。
同时通过PacBio和Nanopore双平台测序数据组装结果的比较发现,利用PacBio数据进行基因组组装Contig N50一般达到Mb级别,而利用Nanopore数据进行基因组组装,Contig N50指标平均水平基本能再提升2倍或者更高,甚至许多物种能达到几十Mb(如百迈客利用Nanopore测序平台组装的水产动物绿鳍马面鲀基因组,Contig N50高达22 Mb)。
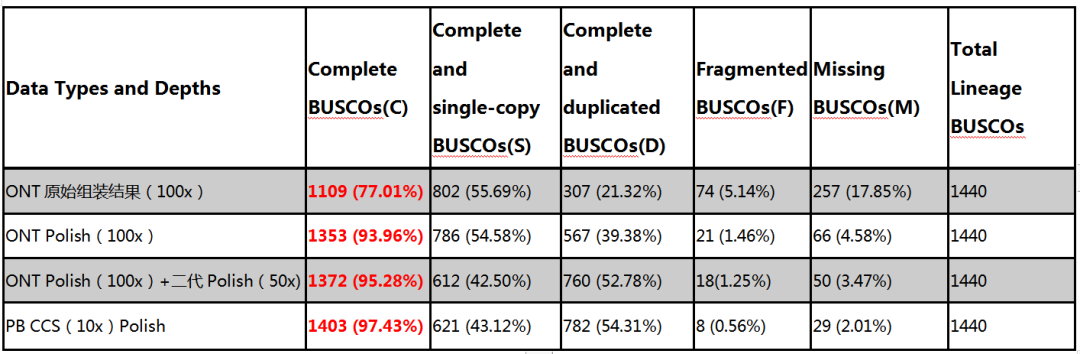
由于Nanopore测序Reads读长长,PacBio Sequel II HiFi模式测序准确性高达99%以上,为了同时利用其双平台各自的优势,我们拟通过Nanopore测序数据对某多倍体植物进行基因组组装,同时通过低深度PacBio CCS数据进行Polish,进而对该多倍体植物基因组连续性,完整性及准确性进行评估,以获得高连续性,高准确的基因组密码信息,测试结果如下:

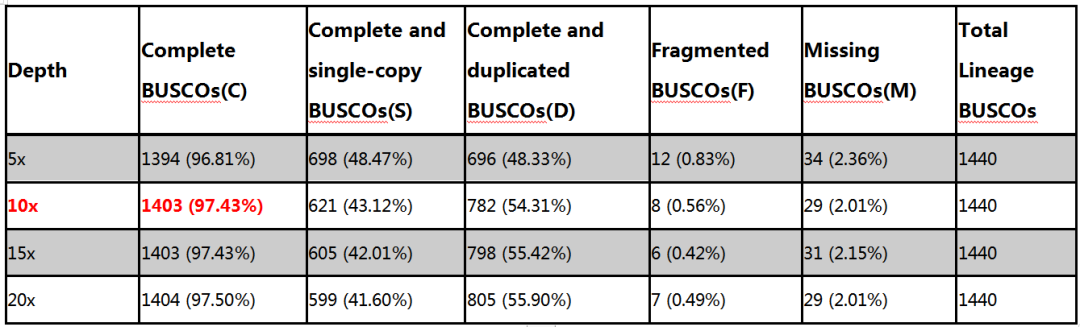
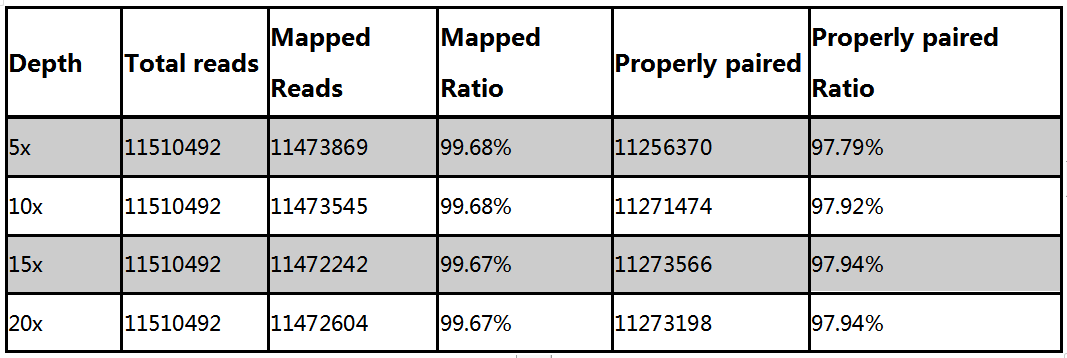
4. 不同深度CCS 数据Polish后二代数据回比结果
5. 通过将20 x CCS数据分别回比到10 x PacBio CCS polish及100 x Nanopore+50 x Illumina Polish后基因组,截取基因组上特性区域,进行组装基因组单碱基准确性的比对与评估,发现10 x PacBio CCS polish后的结果提升效果明显,我们挑选了几个实例如下:
区域1:
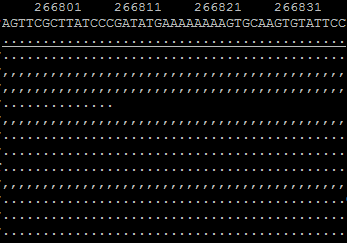
PacBio CCS回比结果(10x CCS Polish基因组)
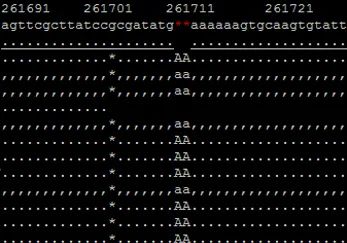
PacBio CCS回比结果(100 x ONT+50 x Illumina Polish基因组)
区域2:
PacBio CCS回比结果(10x CCS Polish基因组)
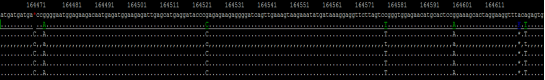
PacBio CCS回比结果(100x Nanopore+50x Illumina Polish基因组)
上述分析结果中,进一步证实了前期的推测,利用Nanopore超长读长的优势,组装获得高连续性基因组(Contig N50 约10 Mb),同时结合PacBio CCS高准确性测序,进一步提升基因组中单碱基的准确度,即鱼和熊掌可兼得。高连续性基因组的获得,对后续功能基因定位,结构变异检测具有重要的意义;同时高准确的基因组的获得,对于超大基因组,多倍体基因组等复杂基因组的LTR的热点区域的研究更具突破性的意义。除此之外。在很多动植物基因组上的确存在高度复杂的区域,即使通过高深度PacBio CCS数据依然无法矫正,这就需要通过其它相应的技术及软件参数整合来提升基因组的准确性。
四、双平台(Nanopore+PacBio)基因组De novo高分文章赏析

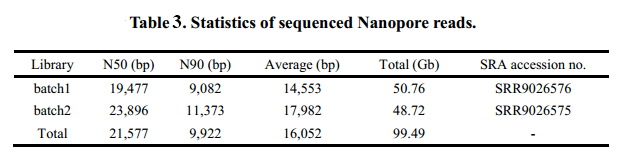
在对同源四倍体紫花苜蓿(Medicago sativa L.)基因研究中,首先利用了70 GB,~22x PacBio CCS数据进行基因组组装,组装获得紫花苜蓿基因组大小3154 Mb,Contig N50=459 kb, 然后利用ALLHiC进行同源染色体组群的划分,最后通过Hi-C互作热图、遗传图谱共线性、ONT数据回比、BUSCO完整性、转录组对基因组完整性等进行评估,值得注意的是在ONT数据回比评估中(Table 3),文中筛选了99 GB ONT long reads中的最长200条reads(ranged from 95 to 263 Kb)进行回比,发现89%的的reads都能比对到single染色体上,结合其它评估方法,进一步说明了组装及染色体位置的准确性。

在小垫柳(Cushion willow)基因组组装中,首先利用SMARTdenovo对纠错后的74xONT数据进行组装,然后分别利用125xPacBio数据(two rounds )与Illumina数据(five rounds )进行polish,基因组完整性评估后,利用Hi-C将Contig挂载到染色体水平,最终组装获得小垫柳基因组大小339.588 Mb,Contig N50=9.522 Mb。 (Table 4)

五、百迈客Nanopore、PacBio平台动植物基因组合作文章总览(部分)
北京百迈客生物科技有限公司自2015年引入Pacbio测序平台,2017年初引入Nanopore测序平台以来,截止到目前百迈客已拥PacBio平台:RS Ⅱ、PacBio Sequel、PacBio sequel Ⅱ;Nanopore 平台:PromethION-48、PromethION-β、Nanopore GridION、MinION,拥有主流三代测序仪,尤其针对复杂超大基因组测序,百迈客生物具有三代测序通量,以满足超大基因组的组装需求。同时PacBio和Nanopore两大主流三代测序平台各自及组合经验,为老师们提供了可参考且全面优质的选择!选择我们,提供专属于您基因组套餐!




 京公网安备 11011302003368号
京公网安备 11011302003368号