

功能富集分析流程是怎样的?
接下来,我们以KEGG数据库为例,带大家梳理下功能富集分析流程:
1、教你怎么将KEGG数据库本地化。
2、如何自己定义功能富集的背景基因集(以物种人类为例,选取一个平台检测的所有基因和全部的人类已知基因做背景基因有什么区别么?差异在哪里?)
3、如何自己写代码实现功能富集核心原理。
小编有话说
首先做一个基因集合的功能富集,必须搞清楚一件事情,基因功能富集和基因功能注释的差别:前者是一个 gene set 是否共同参与完成了一个生物学过程,而功能注释只是针对single gene来说的,这个基因可能参与的生物学过程,有一些什么功能,只需要看这个基因是否在某个通路中出现,一个简单的map过程而已,没有用到统计学模型,不需要显著性计算。
而评价一个gene set是否共同参与完成了某个生物学过程,这有很多统计学模型和算法来评估这个事情,比如最常见的超几何模型和GSEA功能富集的排秩原理。这里小编会以超几何原理为例,告诉你怎么自己写代码实现这个功能富集过程,加深对功能富集的理解。
第一步 KEGG数据库本地化
》获取KEGG通路中所有通路列表
1.1 KEGG官网https://www.genome.jp/kegg/,首先我们看一下KEGG数据库中的Pathway。
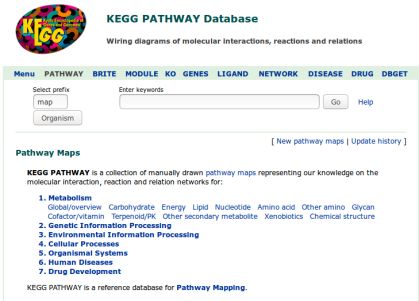
KEGG将pathway分为了七大类,每一大类下面有小类,大家可以自己去详细看看是什么样的。
| 1. Metabolism 代谢 | 代谢 |
| 2. Genetic Information Processing | 遗传信息处理 |
| 3. Environmental Information Processing | 环境信息处理 |
| 4. Cellular Processes | 细胞过程 |
| 5. Organismal Systems | 组织系统 |
| 6. Human Diseases | 疾病 |
| 7. Drug Development | 药物 |
1.2 进入如下这个网址:
https://www.kegg.jp/kegg/rest/keggapi.html#list,点击如图所示链接得到KEGG所有人类通路的列表,截至20190418,收录了330条通路。
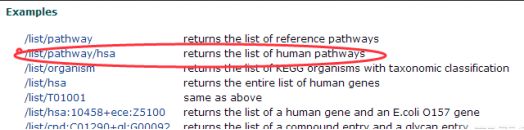
通路列表如下,第一列是KEGG通路ID号,第二列是通路名称。
| path:hsa00010 | Glycolysis / Gluconeogenesis – Homo sapiens (human) |
| path:hsa00020 | Citrate cycle (TCA cycle) – Homo sapiens (human) |
| path:hsa00030 | Pentose phosphate pathway – Homo sapiens (human) |
| path:hsa00040 | Pentose and glucuronate interconversions – Homo sapiens (human) |
| path:hsa00051 | Fructose and mannose metabolism – Homo sapiens (human) |
| path:hsa00052 | Galactose metabolism – Homo sapiens (human) |
| path:hsa00053 | Ascorbate and aldarate metabolism – Homo sapiens (human) |
| path:hsa00061 | Fatty acid biosynthesis – Homo sapiens (human) |
| path:hsa00062 | Fatty acid elongation – Homo sapiens (human) |
1.3 获取任意一个pathway下载网址
进入网址https://www.kegg.jp/kegg/kegg3a.html,点击图片中框框位置,得到通路下载链接:
https://www.kegg.jp/kegg-bin/download?entry=hsa00010&format=kgml

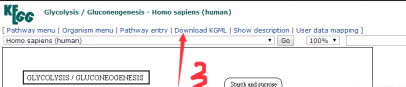
1.4 根据上面下载连接的特点和1.2通路列表id生成所有的KEGG通路下载地址
https://www.kegg.jp/kegg-bin/download?entry=hsa00010&format=kgml
即替换这个网址中entry=hsa00010即可,R代码如下:
# 生成某一通路的链接,最后用迅雷批量下载xml文件
rm(list=ls())
setwd(“/path”)
# 导入kegg文件(通路ID,通路名字)
kegg <- read.table(“KEGG_pathwayID.txt”,header=F,sep=“\t”,quote=“\””,stringsAsFactors=F)
dim(kegg)
# 生成某一通路的链接(手动网页复制)
original_URL <- “http://www.kegg.jp/kegg-bin/download?entry=hsa00010&format=kgml”
replaced_pathwayId <- gsub(“path:”,“”,kegg[,1])
replaced_URL <- sapply(replaced_pathwayId,function(x){gsub(“hsa00010”,x,original_URL)})
hsa_keggURLs <- cbind(pathwayId=replaced_pathwayId,pathwayName=kegg[,2],pathwayURL=replaced_URL)
write.table(hsa_keggURLs,file=“hsa_keggURLs.txt”,sep=“\t”,row.names=F,quote=F)
然后使用wget 命令下载hsa_keggURLs.txt文件中所有通路的xml文件,或者使用迅雷下载也是可以哒!
本期功能富集分析流程先讲到这里,下期我们讲解xml文件的特点和如何根据通路的xml文件提取每个通路中注释的geneid,并获得对应关系。





 京公网安备 11011302003368号
京公网安备 11011302003368号